I was recently reading through the list of ECGs, and came across this one:
What do you think?
This was the interpretation I put into the system:
"Re-entrant wide complex tachycardia with retrograde P waves, slow onset of QRS is c/w either antidromic WPW (ARVT) with pathway insertion in the superior RV, or with VT from the superior RV, most likely RVOT" (Right Ventricular Outflow Tract VT, one of the idiopathic VTs.
Explanation:
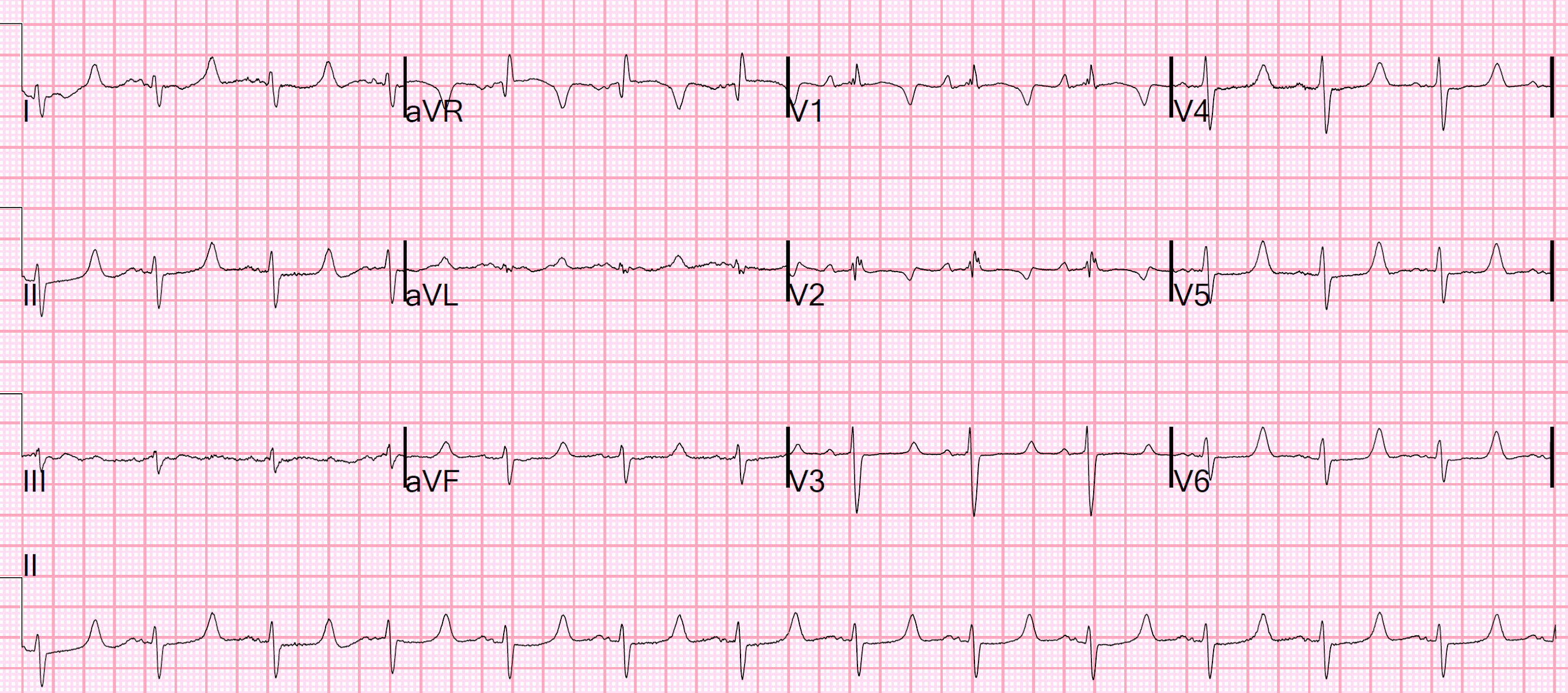
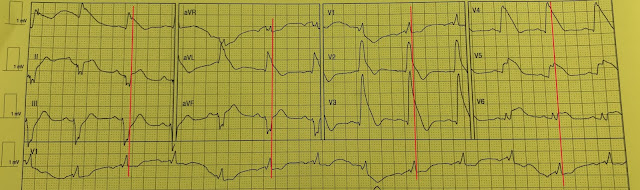
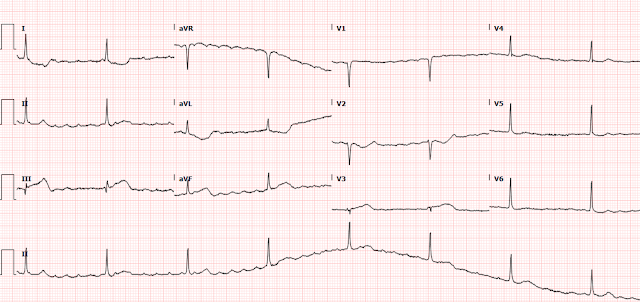
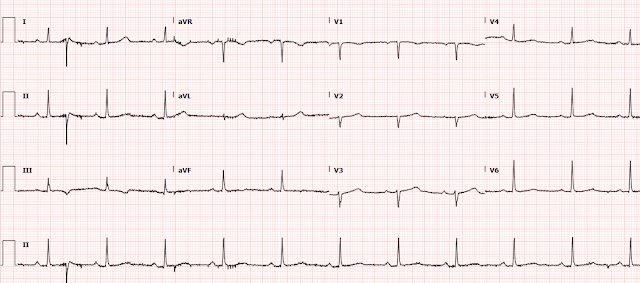
There is a regular wide complex tachycardia at a rate of 138. Here are the important features:
1. V1 and V2 have an LBBB configuration (minimal or no R-wave, deep S-wave)
2. V5 and V6 have an LBBB configuration (monophasic R-wave)
3. Inferior axis (all upright in II, III, aVF)
4. The onset of the QRS is VERY slow (see slow downstroke in V1, V2, slow upstroke in other leads).
This slow onset means that it is either VT or AVRT (Atrioventricular reciprocating tachycardia). AVRT is due to an accessory pathway (similar to WPW, but technically only accessory pathways with baseline delta waves can be called WPW), and when wide it is due to antidromic conduction (down the pathway resulting in pre-excitation of the ventricle -- delta wave with wide QRS). It is the onset of the QRS that is slow -- NOT the latter part.
If this is VT, it is coming from the RV (as there is LBBB pattern). AND it is coming from the superior part of the RV so that the impulse is directed inferiorly (inferior axis). This is typical of Right Ventricular Outflow Tract VT. Other VT that comes from the RV and has LBBB pattern includes ARVD (Arrhythmogenic Right Ventricular Dysplasia, or RV Cardiomyopathy). The latter does not necessarily have an inferior axis.
Apparently, RVOT is also called "Repetitive Monomorphic VT" as it comes in bursts or salvos. At the ver bottom of the post, I have pasted part of an article from UpToDate, my favorite online resource, on this topic.
Here I annotate the ECG to show the retrograde P-waves
Arrows show the middle, or nadir, of the inverted P-wave.
The inverted P-wave is not the upright component of the wave, but the trough.
The blue line is drawn at the onset of the inverted P-waves
Case continued
I looked to find the patient; she was still in the department and this is the story:
A 60-something woman with no cardiac history presented for tooth abscess. Her pulse at triage was 76. The intern elicited a history of chest pain 3 days prior and so ordered an ECG. The intern was unaware of the heart rate at the time of the ECG until it was recorded:
Electrolytes including K and Mg were normal.
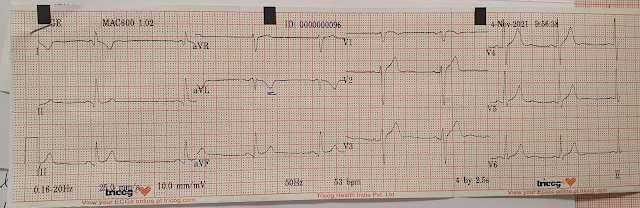
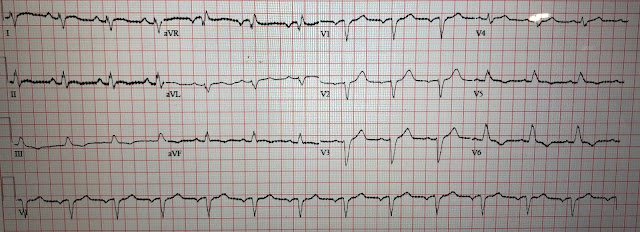
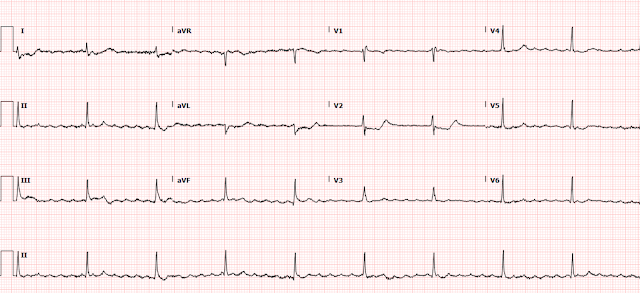
The patient spontaneously converted to sinus rhythm:
Now we see sinus tachycardia with PVCs and otherwise not too remarkable.
What do you think?
My thought on this was that the PVCs have EXACTLY the same morphology as the wide complex tachycardia. This indicates that the irritable focus of the tachycardia is in the ventricle at the exact point of the re-entrant rhythm, and therefore that the first ECG represents VT and not AVRT. The absence of delta waves by itself is supportive but by no means diagnostic, as accessory pathways frequently do not manifest delta waves on the baseline ECG. This is called "concealed conduction."
See comments below by our electrophysiologist on the significance of the PVCs
Again, one does not call it WPW when there is concealed conduction; "WPW" is reserved for those with delta waves. Nevertheless, whether WPW or not, accessory pathways offer another route for AV or VA conduction.
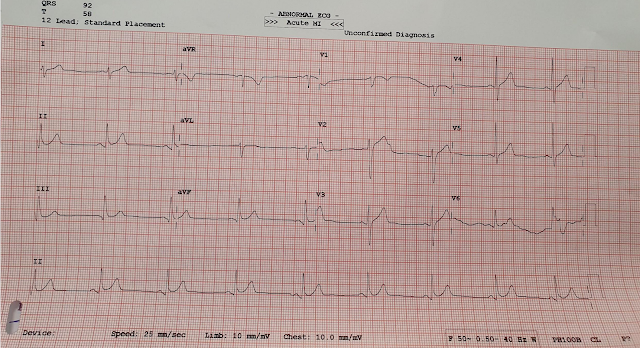
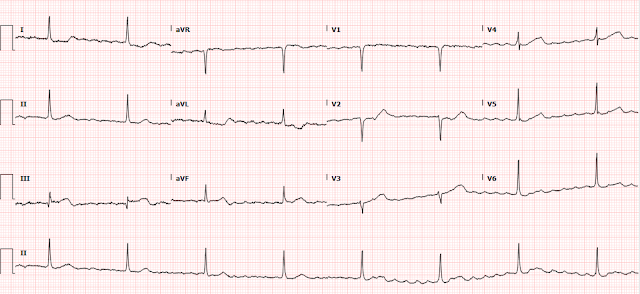
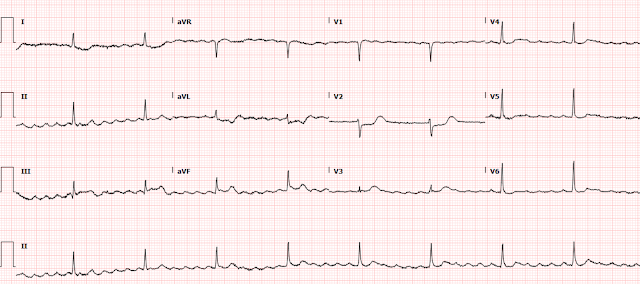
Shortly thereafter, she went back into tachycardia. She was asymptomatic but was hypotensive at 80/50.
Same as before. And it is not spontaneously converting
What do you want to do?
This is almost certainly RVOT, right ventricular outflow tract VT, and this kind of VT is responsive to adenosine!! So if we could trust that she would not revert to VT, adenosine could work. Alas, no short acting treatment such as adenosine or electricity will accomplish the goal of keeping the patient in sinus rhythm.
Repeat: neither electricity nor adenosine will work to convert the rhythm. Neither will solve the problem since we know that she can revert from sinus back to VT, as she has already done it once.
Therefore, a long acting medication or an infusion (or both) are necessary. Given the results of the Procamio Study comparing Procainamide to Amiodarone for stable monomorphic VT (for procainamide, it showed double the efficacy and half the adverse effects), we decided to give procainamide bolus (10 mg/kg over 20 minutes, with the option to give an additional dose of up to 7 mg/kg if necessary) and infusion.
2 g of Mg were also given.
After infusion of only 100 mg, she converted again. She remained in sinus rhythm on the infusion.
Echo:
--Frequent ectopy throughout exam.
--Normal LV size and wall thickness.
--Lower limits of normal left ventricular systolic function with an EF of 52%.
--There is no left ventricular wall motion abnormality identified.
--Normal right ventricular size and function.
--The estimated pulmonary artery systolic pressure is 34 mmHg + RA pressure.
--Based on the appearance of the IVC, the estimated RA pressure is normal.
Outcome:
The patient refused EP study, but the electrophysiologist was certain that this was Right Ventricular Outflow Tract VT (RVOT VT). They discharged her on Diltiazem, which apparently works very well to prevent recurrent bouts of RVOT VT (I was unaware of this and could find no literature anywhere I looked).
Read more here: Idiopathic Ventricular Tachycardias for the EM Physician
Discussion by our Electrophysiologist
Smith: “I thought that the wide complex tachy (WCT) could be AVRT or VT”
EP: "Antidromic AVRT morphology would essentially be the same as “VT” originating from ventricular the insertion site of the accessory pathway. Therefore, traditional criteria for SVT with aberrancy do not apply to antidromic AVRT (except, that negative concordance can never be AVRT!)"
Smith: “But then when the patient converted and had PVCs of exactly the same morphology as the WCT, that it must be VT and not AVRT”
EP: "In cases of intermittent pre-excitation, you could potentially see wide and narrow QRS complex on the same EKG – but those of course will be preceded by P-wave (albeit with short PR interval)"
Smith: “Is this logic supported by evidence? Or is it still likely to be ARVT?”
EP: "Not sure, but PVC’s from RVOT and the wide complex tachycardia of same morphology is highly suggestive of it to be VT.
Another observation on the EKG is that presumably the retrograde atrial activity is quite far out from the next wide complex beat. In other words, if this was AVRT, then you’re assuming that the “PR” interval is very long and is conducting down the accessory pathway. Although there are slowly conducting accessory pathways, I believe they tend to cause Orthodromic AVRT more often. One example is “PJRT” that you might have come across.
On occasion, if someone has “fully pre-excited” beat (i.e., no fusion with AV node), then might be difficult to differentiate from a PVC as “P” wave might just be too close to the onset of QRS – but I don’t recall coming across such situation."
Finally:
In general, if you think a wide complex tachycardia is antidromic AVRT, then it’s “okay” to give AV nodal agent as AV node is still participating in the circuit. That’s whay you might have heard some folks mention using adenosine as diagnostic maneuver even in wide complex tachycardia if patient is hemodynamically stable (if it doesn’t terminate tachycardia, then might elicit AV dissociation if it is VT!). it does cause some vasodilatation, so I wouldn’t do it if someone already is hypotensive.
REPETITIVE MONOMORPHIC VT from UpToDateRepetitive monomorphic VT (RMVT) is characterized by frequent short "salvos" of monomorphic nonsustained VT (waveform 1). It was first described by Gallavardin in 1922 and is variously described in the literature as RMVT, RV tachycardia, RVOT tachycardia, catecholamine-sensitive VT, adenosine-sensitive VT, and exercise-induced VT. Approximately 10 to 15 percent of cases arise from the left ventricular outflow tract [26-28].
Although RMVT is considered to occur in "normal" hearts, static and cine-magnetic resonance imaging often reveal mild structural abnormalities of the RV, primarily involving the free wall (focal thinning, fatty infiltration, and wall motion abnormalities) [13-15]. The functional significance of these changes is uncertain. In the few cases studied, DNA from myocardial biopsies of ventricular muscle has been normal [29].
Epidemiology and clinical features — RMVT occurs almost exclusively in young to middle-aged patients without structural heart disease [1,2,4-9]. There has generally been no predilection on the basis of sex, although a 2:1 female predominance was observed in one report [26]. A surprising number of competitive athletes (particularly cyclists) are identified in many series of RMVT.
The most common associated symptoms are palpitations and lightheadedness during episodes [5,6]. In one illustrative report of 18 patients, twelve had symptomatic arrhythmia, two of whom had syncope, and six were completely asymptomatic.
Most arrhythmias are nonsustained (usually 3 to 15 beats), but up to one-half of patients have some sustained episodes, and some patients have only sustained VT [6,9,13,30]. (See 'Paroxysmal sustained VT' below.)
Bursts of nonsustained VT are typically provoked by emotional stress or exercise, often occurring during the "warm-down" period after exercise, a time when circulating catecholamines are at peak levels [5,6,8]. There may also be a circadian pattern, with prominent peaks between 7 and 11 AM and 4 and 8 PM, correlating with periods of increased sympathetic activity [31]. In some patients, a critical "window" of heart rates (upper and lower thresholds) that result in occurrence of the arrhythmia can be defined [27].
The inducibility of RMVT by stress or catecholamine infusion is suggestive of an abnormality in cardiac sympathetic function. Consistent with this hypothesis is evidence of regional cardiac sympathetic denervation in some patients with RMVT and structurally normal hearts (five of nine compared with zero of nine controls in one report) [32]. Patients with RMVT may also have regions of impaired neuronal reuptake of norepinephrine, leading to increased local synaptic catecholamine concentrations and downregulation of myocardial beta adrenergic receptors [33].
There may also be sex-specific triggers. In a report of 47 men and women with RMVT, states of hormonal flux (premenstrual, gestational, perimenopausal, administration of birth control pills) were the most common trigger for RMVT in 59 percent of women and were the only recognizable triggers in 41 percent [34]. Men were more likely than women to identify exercise, stress, or caffeine as a trigger (92 versus 41 percent).
Site of origin — RV tachycardias usually originate from the septal aspect of the RVOT [6,26,28,35-38]. A nine site mapping schema of the septal RVOT has proven useful in localizing RVOT tachycardias on the basis of their 12 lead ECG morphology (figure 1) [39].
RV tachycardias typically arise from a very narrow area just inferior to the pulmonary valve in the anterior aspect of the RVOT [37]. Endocardial mapping in such patients shows that the earliest site of endocardial activation occurs in this region [6,9].
Less commonly, sites of origin have been mapped to the RV inflow tract, the free wall of the RVOT, the root of the pulmonary artery, the left and right aortic sinus of Valsalva, the left ventricle, the mitral annulus, and the papillary muscles [26,36,38,40-49].
Electrocardiographic features — The typical rate of RMVT ranges from 140 to 180 beats/min, and may fluctuate based upon catecholamine levels. The VT cycle length often prolongs prior to termination.
RV outflow tract — The majority of RMVT episodes have a characteristic ECG appearance with two main features [1,2,4-9,39]:
●Left bundle branch block
●Inferior axis
This morphology is consistent with the RVOT origin seen by catheter ablation and endocardial mapping [6,37]. This ECG "signature" accounts for at least 70 percent of all idiopathic VTs [1].
The ECG pattern of RV tachycardia initiation may provide information about the site of origin and the arrhythmogenic mechanism as illustrated by the following observations:
●In a series of 32 patients with exercise-induced RMVT, VT usually began without a change in cycle length. Arrhythmias that initiated in this manner had an inferior axis, and appeared to be related to triggered activity due to delayed afterpotentials [50]. By comparison, VT initiated with a long-short sequence was more often nonsustained and often had a superior axis, suggesting an origin in the body or septal region of the ventricle; the mechanism for this VT is probably early afterpotentials.
●In a report of 14 patients, those with septal compared with free wall sites were significantly less likely to have notching of the QRS complex (29 versus 95 percent) and more likely to show early precordial transition by lead V4 (79 versus 5 percent) [51]. In addition, a positive R wave in lead I distinguished posterior from anterior septal and free wall sites.
●The degree of similarity of 12-lead ECG waveforms between VT and a pace map can be used to estimate the likelihood of successful ablation at that site. (See 'Radiofrequency ablation' below.)
The majority of patients with RV tachycardia have a single ECG morphology [23]. However, occasional patients present with multiple tachycardia morphologies arising from discrete sites in the RVOT [52]. The presence of multiple left bundle VT morphologies, particularly during EP study, should suggest the possibility of ARVC [23,53,54]. (See 'Distinction from ARVC' above.)
LV outflow tract — Electrocardiographic criteria have also been described for RMVT originating in the LVOT [26,27]. In a series of 33 patients with RMVT, four (12 percent) had LV sites of origin that could be predicted by two patterns [26]:
●A right bundle, inferior axis morphology with a monophasic R wave in V1 that arose from the left fibrous trigone (waveform 2).
●A pattern similar to typical RMVT from the RVOT (left bundle, inferior axis) except that the precordial transition was earlier (at V2 for the LVOT as compared with V3 or later for the RVOT).
Other reports have characterized unique QRS morphologies for idiopathic VT arising from the sinuses of Valsalva [9,43,44]. Although there is some interindividual variability, premature ventricular complex/contraction (PVC; also referred to a premature ventricular beats or premature ventricular depolarizations) arising from the left aortic sinus tend to be negative in lead I and have a "w" pattern in V1, while PVC with a broad R wave in V1 is characteristic of a right aortic cusp origin. The precordial R wave transition is much earlier when VT originates from either aortic sinus of Valsalva compared with the RVOT, since the LVOT is posterior to the RVOT [44].
Electrophysiologic features — RMVT can be induced in the EP laboratory, although usually not with programmed stimulation [5,6,23,27,55]. In most patients, sustained or nonsustained episodes occur in response to burst atrial or ventricular pacing, and are greatly facilitated by isoproterenol or epinephrine infusion [5,6,27,30,55].
These electrophysiologic observations suggest that triggered activity due to delayed afterpotentials, rather than reentry, is the mechanism of RMVT. The response to "pharmacologic probes" further strengthens this hypothesis. RMVT has been terminated with adenosine, verapamil, and beta blockers, all of which interfere with the cAMP-mediated slow inward calcium current [29,40,56-58].
These observations are consistent with the hypothesis that RMVT results from triggered activity induced by cAMP-mediated delayed after depolarizations (DADs) [30,59]. The increase in cAMP activity may, at least in some patients, result from an acquired somatic cell mutation in the inhibitory G protein G-alpha-i2 at the site of the arrhythmogenic focus [59,60].
However, the lack of specificity of these probes and the absence of a uniform response supports the general consensus that the mechanism of RMVT is incompletely characterized and may vary among individuals. Additional support for other mechanisms is based upon the observation that the tachycardia may, in some patients, terminate with overdrive pacing, ventricular extrastimulation, or autonomic modulation using Valsalva maneuver or carotid sinus pressure [9].
EP studies may also help distinguish RMVT occurring in the absence of structural heart disease from that in ARVC [23,24]. (See 'Distinction from ARVC' above.)
Prognosis — The prognosis of RMVT is almost uniformly good [4-9,55]. The following observations from early studies illustrate the range of findings:
●Two initial series evaluated 30 and 18 patients [5,6]. The arrhythmia responded to a variety of antiarrhythmic drugs, including type I drugs and propranolol. At a mean of 30 months in one study and a range of 0.5 to 8 years in the other, there were no deaths or episodes of cardiac arrest [5,6].
●A third report consisted of 24 young patients, most of whom had RV tachycardia and two-thirds of whom were symptomatic [7]. At a mean follow-up of 7.5 years, three patients died suddenly; none was taking antiarrhythmic drugs at the time. The SCD events may have been due to RMVT itself, although patients with other syndromes now known to be malignant may have been included (eg, Brugada syndrome, arrhythmogenic right ventricular cardiomyopathy). Alternatively, a tachycardia-induced cardiomyopathy may have predisposed patients to additional arrhythmias. (See "Arrhythmia-induced cardiomyopathy".)
Malignant variant — More recent studies in which these other syndromes were unlikely have identified a malignant variant of RMVT. Polymorphic VT and VF, which are malignant arrhythmias, have been demonstrated in patients with RMVT [61,62]. In one series, three patients with RMVT later developed VF. In these patients, PVCs were more closely coupled to prior beats than is usual for RMVT [61]. It was postulated that relatively early triggered beats occurred in a vulnerable period during repolarization, resulting in VF.
In a subsequent report, 16 patients with frequent PVCs from the RVOT were noted to have polymorphic VT or VF that was initiated by one of the PVCs [62]. In contrast to the first study, the coupling interval of the PVCs in patients who developed malignant arrhythmias was not different from that in 85 other patients with RMVT who did not have malignant arrhythmias. Radiofrequency (RF) ablation successfully eliminated the RVOT PVCs in 13 patients and modified the PVCs in the other three. Over a mean follow-up of 54 months, none had recurrent VF or syncope. (See 'Radiofrequency ablation' below.)
The prevalence of this malignant variant, and whether it represents a distinct disorder from RMVT, is unclear. The high frequency in the above study (16 of 101 patients) is probably a substantial overestimate due to referral bias. In addition, it is possible that some cases categorized as polymorphic VT were simply the common form of RVOT VT in which QRS morphology varied during the tachycardia due to fluctuations in loading conditions. An editorial accompanying this report addressed the issue of whether RF ablation should now be considered in all patients with RMVT [63]. Due to the relatively high prevalence of RVOT PVCs, the rarity of malignant RVOT VT/VF, it is reasonable to focus concern on patients with the following higher risk characteristics:
●A history of syncope
●Very fast VT (>230 beats/min)
●Very frequent ectopy (>20,000 PVCs/day)
●PVCs with a short coupling interval
Treatment of RMVT — Therapeutic decisions for RMVT should consider that many patients are young and otherwise healthy. As a result, ablative therapy may be preferable to chronic administration of antiarrhythmic drugs.
Medical therapy — Medical therapy serves two roles in RMVT: termination of the arrhythmia; and prevention of recurrence. RMVT can be terminated with adenosine and beta blockers, all of which interfere with the cAMP-mediated slow inward calcium current [29,40,56-58].
For prevention of recurrence, beta blockers are often used as first-line agents. These drugs are attractive since their side effect profiles are mild in comparison with antiarrhythmic agents [64].
Propranolol has prevented recurrence in as many as 14 of 22 patients with a typical RVOT origin of RMVT [6,65]. However, other studies have found that these agents were much less likely to prevent recurrent RMVT, although the combination of a beta blocker with a class I drug may be useful [9,55].
Class I antiarrhythmic agents (table 2) alone are helpful in some patients. However, class III drugs (sotalol and amiodarone) may be preferred, especially in patients with arrhythmia that is refractory to other drugs [9].
Radiofrequency ablation — Due to the limited efficacy and potential side effects of antiarrhythmic drugs, there has been increasing use of radiofrequency (RF) ablation in patients with symptomatic RMVT. Professional society guidelines for the management of ventricular arrhythmias and the prevention of SCD indicate that there is evidence and/or general agreement supporting RF ablation in patients with symptomatic idiopathic VT that is drug-refractory, or in such patients who are intolerant of drugs or do not desire long-term drug therapy [64].
The 2019 HRS/EHRA/APHRS/LAHRS Expert Consensus Statement on Catheter Ablation of Ventricular Arrhythmias recommended catheter ablation in the following patients with idiopathic VT and without structural heart disease [66,67]:
●Severely symptomatic patients with monomorphic VT.
●Monomorphic VT in patients in whom antiarrhythmic drugs are not effective, not tolerated, or not desired.
●Patients with recurrent sustained polymorphic VT and VF (electrical storm) that is refractory to antiarrhythmic therapy when there is a suspected trigger that can be targeted for ablation.
Success rates for RF catheter ablation range from 80 to 100 percent [28,35,36,40,41,68]. The success rate depends in part upon the location of the focus; the success of catheter ablation for idiopathic VTs in atypical positions is generally not as high as for RVOT locations [36]. The degree of similarity of 12-lead ECG waveforms between VT and a pace map can be used to estimate the likelihood of successful ablation at that site [69]. A mean absolute deviation >12 percent suggests sufficient dissimilarity to dissuade ablation at that site.
Although successful ablation of LVOT tachycardia has been performed using an endocardial approach, coronary venous mapping and percutaneous approaches to the pericardial space have shown that some of these VTs arise from the LV epicardium [28,45,46,68,70]. There have also been several reports of successful ablation of RMVT from the left and right sinuses of Valsalva [43].
Radiofrequency ablation is generally associated with a low rate of procedural complications [28,35-38,40]. (See "Overview of catheter ablation of cardiac arrhythmias", section on 'Complications'.)
Long term follow-up of patients successfully treated with radiofrequency catheter ablation is limited. In two reports of 42 patients in whom all tachycardias were successfully ablated, only five (12 percent) had a detected recurrence during a 2 to 50 month follow-up [68,71].
The likelihood of successful ablation may be less when the site of origin is not endocardial or not definitively identified during mapping [68]. In a review of 75 patients with presumed RVOT VT, the inability to identify a focus, and therefore the success rate, correlated with the QRS duration in lead V2 [72]. The success rate was 95 percent when the QRS duration in V2 during pace mapping was ≥160 ms in duration compared with only 54 percent when the QRS duration was <160 ms. Left-sided and epicardial ablation strategies were not pursued in this experience.
Epicardial ablation — A possible explanation for failed radiofrequency ablation is that the arrhythmia arises from an epicardial rather than an endocardial focus. In one report of failed ablation (either acute failure of the procedure or late recurrence of arrhythmia) in 30 patients with VT (15 with apparently normal hearts), subxiphoid instrumentation of the pericardial space was used for both epicardial mapping and ablation [70]. Twenty-four of the VTs appeared to originate from the epicardium; 17 were successfully ablated, while the other seven had sites that were inaccessible primarily due to interference from the left atrial appendage. Six of these seven patients could be ablated from the left coronary cusp.
PAROXYSMAL SUSTAINED VTParoxysmal sustained VT is another clinical syndrome of idiopathic right ventricular tachycardia that resembles RMVT in many ways [6,9,13,73]. The most frequent QRS morphology is a left bundle branch block with an inferior axis, and the typical site of origin is the superior septal aspect of the RVOT. Furthermore, patients with paroxysmal sustained VT appear to respond to antiarrhythmic agents, particularly adenosine, in a manner similar to patients with RMVT, although data are limited [73].
Because of these similarities, it is not clear if paroxysmal sustained VT is a distinct clinical syndrome. Compared with nonsustained RMVT, this disorder is more often symptomatic and less often exercise-provoked. It is also more frequently induced by programmed stimulation, although isoproterenol may facilitate induction in some cases.
On the other hand, some investigators dispute the existence of paroxysmal sustained VT as a syndrome distinct from RMVT. One study, for example, found that 58 percent of patients with RMVT had at least one episode of sustained VT [9]. In addition, electrophysiologic studies may reproduce sustained VT in patients who present with only nonsustained VT (particularly during isoproterenol infusion) or vice versa. It has also been suggested that the incidence of sudden cardiac death in RMVT is actually due to overlap with paroxysmal sustained VT. For these reasons, the two syndromes are occasionally considered together as idiopathic right ventricular tachycardia, or simply as RMVT.