
Written by Pendell Meyers
A middle aged adult presented with acute undifferentiated chest pain.
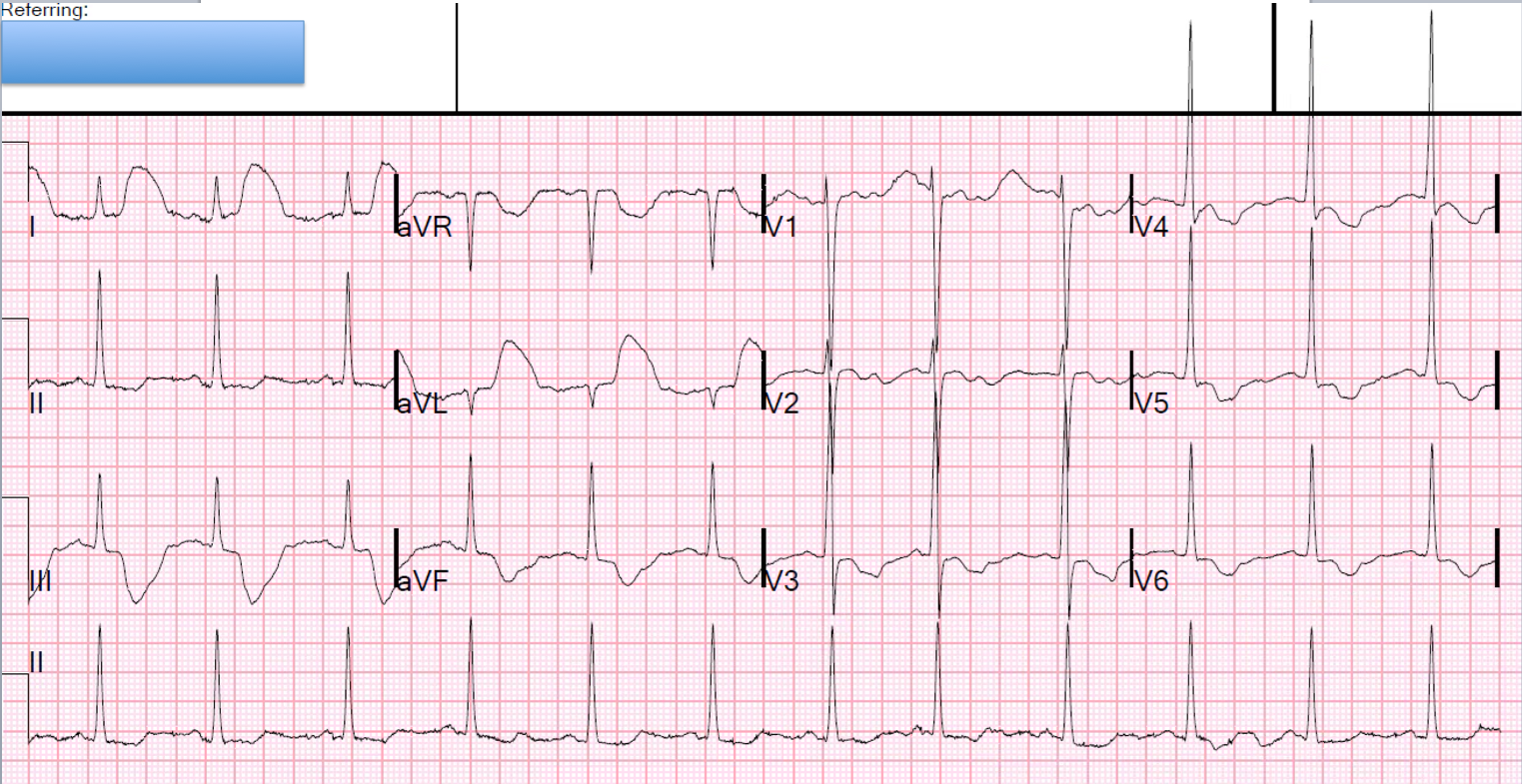
Here is his ECG at triage:
.JPG) |
| What do you think? |

The cath lab was deactivated.
All troponins were undetectable.
No other significant pathology was found upon further chest pain workup.
It seems almost certain to me that the first ECG does represent artifact simulating OMI, which I believe fits with the pattern of arterial pulse tapping artifact.
Check out this case of similarly appearing arterial pulse tapping artifact:
Bizarre (Hyperacute??) T-waves
See more info on arterial pulse tapping artifact:
Arterial pulse tapping artifact
https://www.aclsmedicaltraining.com/blog/guide-to-understanding-ecg-artifact/
This online article references the article below by Emre Aslanger, one of our co-editors:
Aslanger E, Yalin K. Electromechanical association: a subtle electrocardiogram artifact. Journal of Electrocardiology. 2012;45(1):15-17. doi:10.1016/j.jelectrocard.2010.12.162.
Incredibly, this case was just published in Circulation on January 22, 2018 (thanks to Brooks Walsh for finding this!)
Asymptomatic ST-Segment–Elevation ECG in Patient With Kidney Failure. https://doi.org/10.1161/CIRCULATIONAHA.117.032657. Circulation. Originally published January 22, 2018
Here is a case from Circulation year 2000 that was misdiagnosed as due to pancreatitis. But you can tell from the normal lead III that this was a right arm electrode problem:
http://circ.ahajournals.org/content/101/25/2989.full
===================================
MY Comment, by KEN GRAUER, MD (8/26/2022):
===================================
Today’s case provides an excellent example of how the 1st time you see an ECG phenomenon — it may pass unrecognized. But after one learns about the phenomenon — it becomes EASY to recognize in the future!
- The clinicians who initially saw today's patient were fooled by what superficially looks like ST elevation in high lateral leads I and aVL — and — with what looks like marked ST depression in virtually all other leads.
KEY Point: Artifact is deceptively common in clinical practice. The BEST way not to overlook artifact — is to be aware of how common it actually is! I’ll add the following points regarding CLUES to today’s case:
- CLUE #1: The 1st thing to notice about the initial ECG ( = ECG #1 that I've reproduced in Figure-1 below) — is that there is a tremendous amount of baseline artifact. This is especially true in the limb leads — where none of the seemingly elevated and depressed ST segments look the same. In the long lead V1 rhythm strip — each of the 10 beats manifest a different variation on the shape of the ST segment. When there is much artifact elsewhere on a tracing — the chances increase greatly that artifact is also affecting the ST segments you are concerned about.
- CLUE #2: The shape of the elevated and depressed ST segments in the limb leads is bizarre. The ST segment is jagged in the 2 limb leads with ST elevation (ie, leads I, aVL) — and the deepest part of the depressed ST segments in leads III and aVF is almost pointed. This does not look physiologic. In general, when ECG deflections look bizarre and “unphysiologic” — there is an excellent chance that such deflections are not real!
- CLUE #3: The shape of the depressed ST segments in all 6 chest leads looks very similar (ie, with a "rounded scoop", showing approximately the same amount of ST depression in each of these 6 leads — as highlighted in GREEN). The presence of geometric shapes (in this case, the "rounded scooping") — is also unlikely to be physiologic.
- CLUE #4: All of the above described unusual shapes (ie, the peak of the ST elevation in leads I,aVL — the negative peaking in leads III and aVF — and the lowest part of the rounded GREEN scoopings) — occur at a fixed interval with respect to the preceding QRS complex. This suggests us that whatever is producing these deflections must be related to cardiac contraction (and/or to arterial pulsation)!
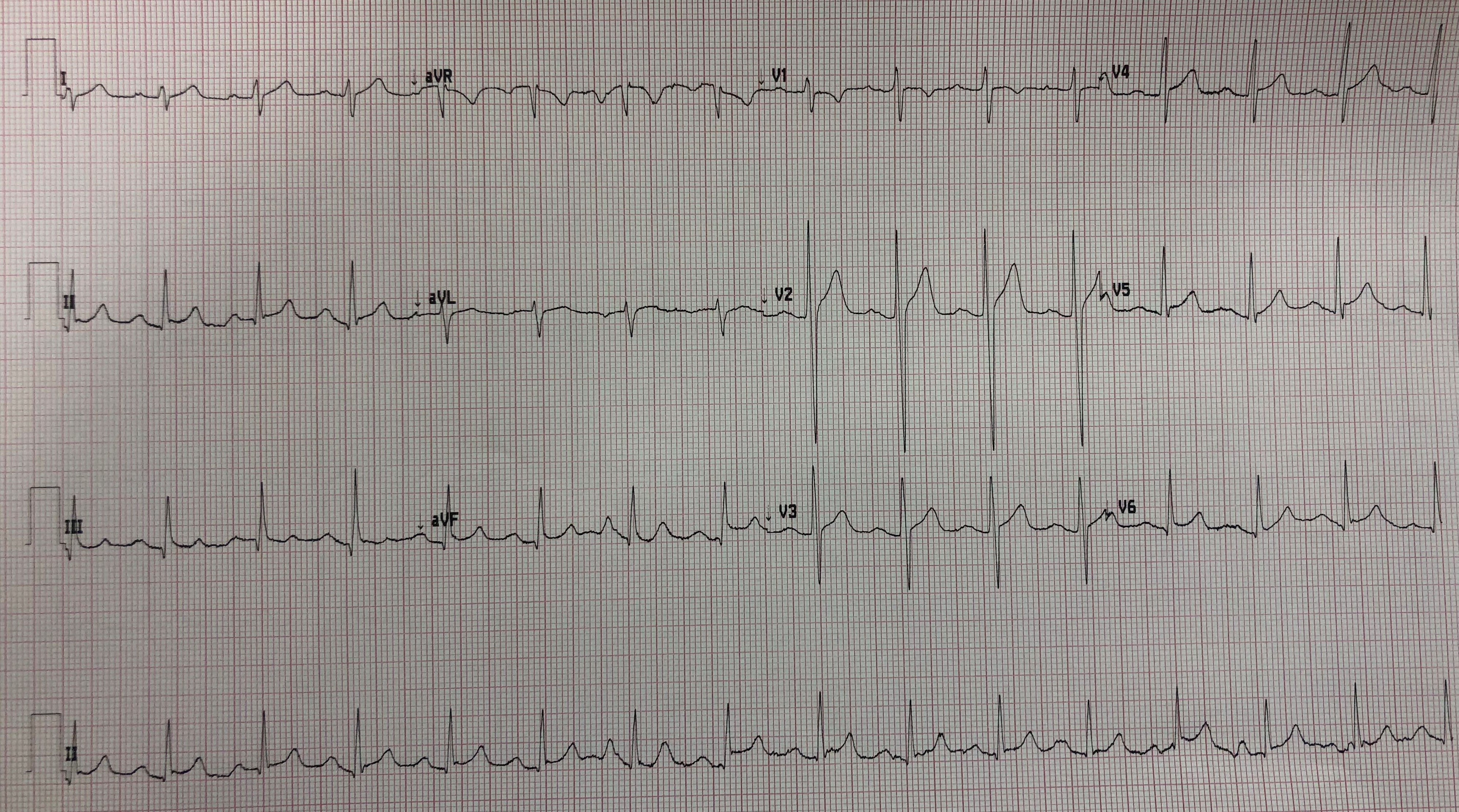
- But it is CLUE #5 — that clinched the diagnosis of artifact for me (See below Figure-1).
-USE.png) |
| Figure-1: The 2 ECGs from today's case. I've labeled the artifact in ECG #1 (See text). |
CLUE #5: The distribution in ECG #1 of the bizarre ST-T wave deflections precisely follows the location and relative amount of amplitude distortion predicted by Einthoven’s Triangle.
- The amount of artifactual ST segment deviation is approximately equal in 2 of the standard limb leads (ie, outlined in RED in leads I and III) — and, not seen at all in the 3rd standard limb lead (ie, the ST segment is neither elevated nor depressed in lead II). By Einthoven’s Triangle (See Figure-2) — the finding of equal ST segment amplitude artifact in Lead I and Lead III, localizes the "culprit" extremity to the LA ( = Left Arm) electrode.
- The absence of ST elevation or depression in lead II is consistent with this — because, derivation of the standard bipolar limb lead II is determined by the electrical difference between the RA ( = Right Arm) and LL ( = Left Leg) electrodes, which will not be affected if the source of the artifact is the left arm.
- By Einthoven's Triangle — the finding of maximal amplitude artifact in unipolar lead aVL confirms that the left arm is the “culprit” extremity (highlighted in RED in lead aVL).
 |
| Figure-2: Use of Einthoven's Triangle to determine the electrical voltages in the 3 standard limb leads. |
NOTE: I reproduce below in Figures 3, 4 and 5 — the 3-page article by Rowlands and Moore (J. Electrocardiology 40: 475-477, 2007) — which is the BEST review I’ve seen on the physiology explaining the relative size of artifact amplitude deflections when the cause of the artifact is a single extremity. These principles are illustrated by the colored deflections that I drew in the initial ECG that I show above in Figure-1:
- As noted by the equations on page 477 in the Rowlands and Moore article: i) The amplitude of the artifact is maximal in the unipolar augmented electrode of the “culprit” extremity — which is lead aVL in Figure-1 (RED outline of the elevated ST segment in this lead); and, ii) The amplitude of the artifact in the other 2 augmented leads (ie, leads aVR and aVF) is about 1/2 the amplitude of the artifact in lead aVL (BLUE outline of the depressed ST segments in leads aVR and aVF).
- Similarly — the amplitude of the artifact deflections in the 6 unipolar chest leads in Figure-3 is also significantly reduced (to ~1/3 size) from the maximal amplitude seen in leads I, III and aVL (GREEN outline of the scooped ST segments in each of the 6 chest leads).
BOTTOM LINE: You will see artifact frequently in real-life practice. Awareness of the above CLUES facilitates recognizing with 100% certainty that the bizarre ST segment deviations seen on a tracing like the one in today's case are the result of artifact — and are related to arterial pulsations in one of the extremities.
- Nothing else shows fixed relation to the QRS complex in the mathematical relationships described above, in which there is equal maximal artifact deflection in 2 of the 3 limb leads (with no ST segment deviation in the 3rd limb lead) — in which maximal artifact in the unipolar augmented lead will be seen in the extremity electrode that shares the 2 limb leads that show maximal artifact (as according to Einthoven’s Triangle).
- ECG #2 was recorded just minutes after ECG #1 — after repositioning all electrode leads. In your mind's eye — Wouldn't ECG #1 look like ECG #2 if we took away the artifactual deflections highlighted in RED, BLUE and GREEN?
 |
| Figure-3: Page 475 from the Rowlands and Moore article referenced above. |
 |
| Figure-4: Page 476 from the Rowlands and Moore article referenced above. |
 |
| Figure-5: Page 477 from the Rowlands and Moore article referenced above. |






-USE.png)
-USE.png)
-USE.png)



