Coronary thrombosis (twice in the same patient!!) without a stenosis or even a culprit
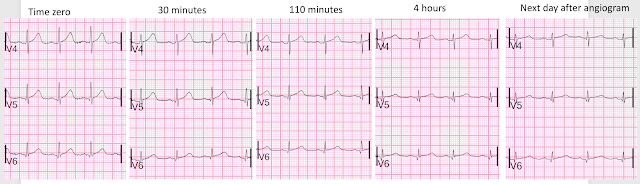
Do not miss the last image at the bottom that shows the series of T-waves in V4-V6
I recently had a discussion with an incredibly smart and fantastic ECG and Cardiology expert. He was skeptical that you can have OMI with Wellens waves without having a major stenosis on angiogram.
I told him I've seen it on occasion and that this happens due to thrombosis of non-obstructive lesions that lead to complete occlusion but that then completely lyse and do not show stenosis by the time of the angiogram. I said that even if there is not a stenosis, they often show a culprit (ulcerated plaque), but not always.
The day after that discussion, this case came in: Transient Occlusion MI (Transient OMI) that occurred twice, but without any stenosis or even a culprit.
The case demonstrates how carefully you must read the ECG, and how carefully you must compare T-waves size. It demonstrates that you must look not only at the angiogram, but to the symptoms, ECG, troponins, and echo for the diagnosis.
You will note that the angiogram on the first presentation manifested an initially unseen occlusive culprit, which was only later seen on angiogram over one year later.
Case
A 40-something woman with DM presented with substernal chest pain that felt like her previous myocardial infarction, 1.5 years earlier. That previous acute MI was diagnosed as MINOCA (Myocardial Infarction with Nonobstructive Coronary Arteries.)
We will give some details on that previous infarction below.
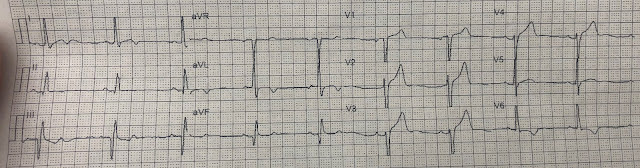
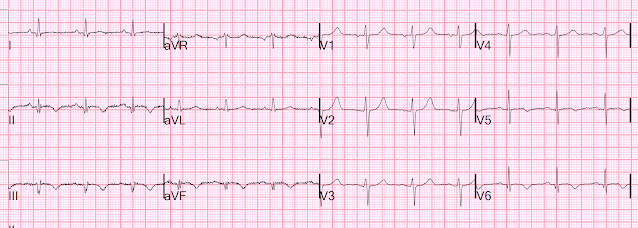
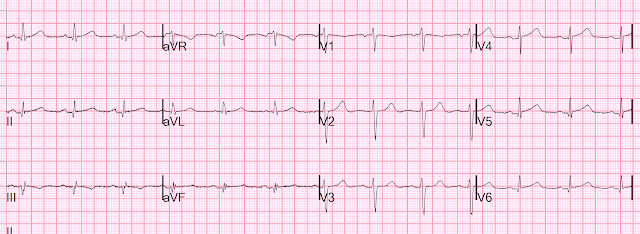
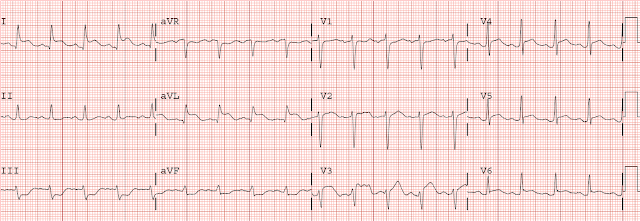
Here is her initial ED ECG (ECG 1):
What do you think?
Smith interpretation: the T-waves in V4-V6 are hyperacute. These are NOT normal T-waves. They are large and fat. The inferior leads look very suspicious as well, with Q-waves and a straight ST segment in leads II and aVF. This is very suspicious as well.
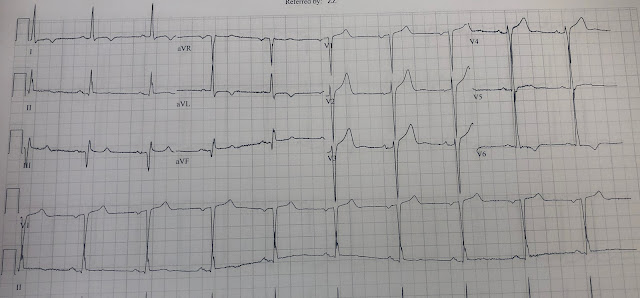
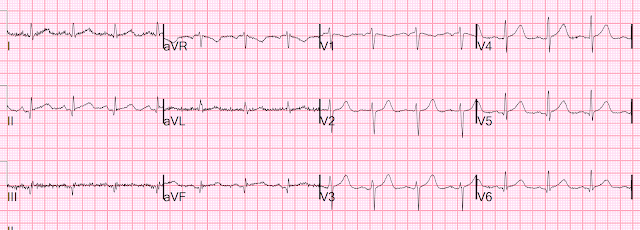
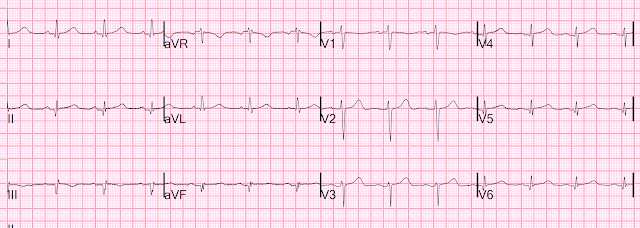
Let's look at her last ECG, just before discharge after hospitalization for the previous MI:
This is completely typical evolution of an inferior Occlusion MI (OMI). There are inferior Q-waves, slight ST elevation ("coving" or Pardee T waves) with T-wave inversion. There are also inverted T-waves in the lateral leads V4-V6. So this previous OMI was inferolateral (I will show the presentation ECG below)
So today's ECG has new upright T-waves in inferior and lateral leads. Is this pseudo-normalization?
No!! The natural evolution of inverted T-waves after acute MI is to become upright over weeks to months. Even without a new MI, one would expect the T-wave in II, III, aVF and V4-V6 to be upright (but not hyperacute, as here!)
True pseudonormalization happens within days and is due to acute re-occlusion of a previous occluded and reperfused coronary.
__________
See these 3 cases for LAD pseudonormalization:
__________
The only way you can tell that ECG 1 above has ischemia is to recognize the hyperacute T-waves. The old ECG only helps you by showing that the Q-waves are old.
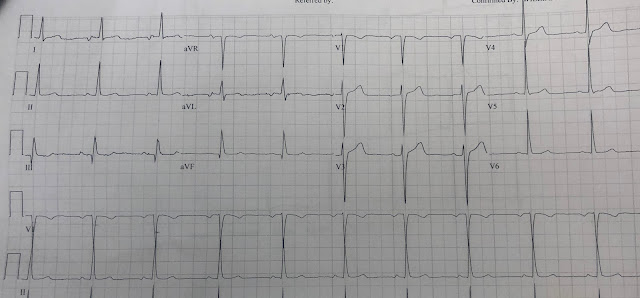
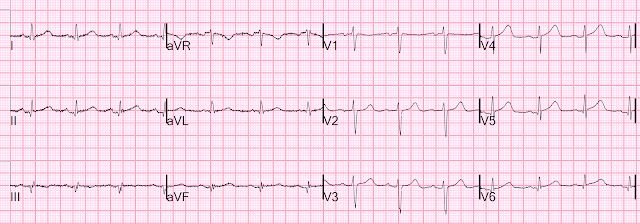
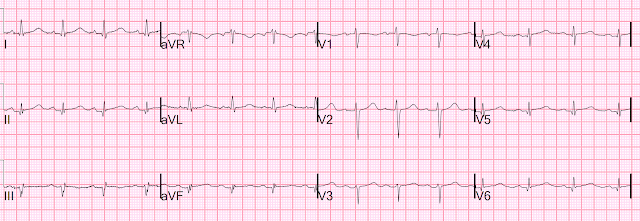
For the prior OMI, here is the presentation ECG:
Inferior, posterior, lateral Occlusion MI that also meets STEMI criteria in inferior leads (STEMI)
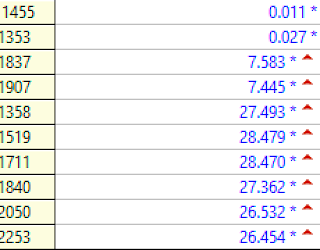
Troponin I profile (contemporary, not high sensitivity) on that previous visit, in ng/mL:
A large acute Occlusion MI
The angiogram at that time showed:
1. Left main: no significant stenosis.
2. LAD: type II vessel that just reaches around the apex. It supplies two diagonal branches. There are luminal irregularities in the LAD but no significant stenosis. Likewise, there is moderate up to 50% disease in D1.
3. LCX: non-dominant. It supplies several tiny OMs and then a large OM. There are no significant stenoses.
4. RCA: It supplies an RPDA and a small RPLA. There are no obvious stenoses or vessel occlusions.
In other words: Non-Obstructed Coronaries
Formal echocardiogram:
Regional wall motion abnormality-lateral and inferolateral
So this previous visit was MINOCA.
Let's get back to the 2nd presentation. Again, ECG 1:
Her pain began to wane and then resolve, and more ECGs were recorded:
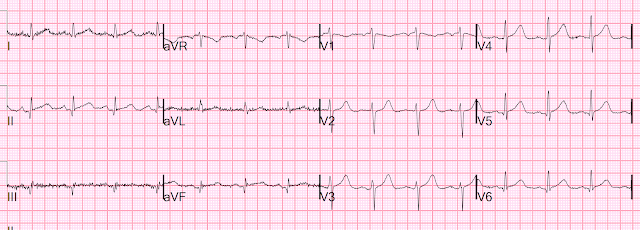
Time 30 minutes:
Lateral T-waves are getting smaller in V4-v6
Time 110 minutes:
V4-V6 T-waves smaller still
Inferior T-waves are inverting ("inferior Wellens' waves, or reperfusion T-waves)
Time 4 hours
Now there is complete resolution of the hyperacute T-waves and inversion of inferior T-waves.
However, no one noticed that these T-waves are ischemic, as far as I can tell. In fact, later interpretations were "She has no current of injury on her EKGs."
The first high sensitivity troponin I was 185 ng/L (this number has a 70% positive predictive value for type 1 MI in our department, but not necessarily for Occlusion MI).
Subsequent trops went to 860, then 2401, then 6658 ng/L.
Since the pain was resolved, and the ischemia was never recognized on the ECG, she was put on heparin and aspirin and angiography was delayed until the next day.
Angiographic findings:
1. Left main: No stenosis.
2. LAD: tortuous. Moderate diffuse disease distally toward the apex. No
obvious focal stenosis. Supplies a diagonal branch without stenosis.
3. LCX: non-dominant. Supplies very small OMs and a medium sized tortuous
LPLA with a very distal 80% stenosis in a small caliber branch that, upon
review of her previous angiogram, had been occluded and is now open.
4. Ramus intermedius: no significant stenosis.
5. RCA: dominant. Supplies an RPDA and small RPLA. No stenosis.
In other words: Non-Obstructed Coronaries
To me, this implies that the PREVIOUS diagnosis of MINOCA was incorrect, as the angiographer now sees an artery that was closed at the previous angiogram but is open now.
But this 2nd angiogram does not show a culprit for the 2nd visit, as far as I can tell.
It is unclear to me if this same vessel is the culprit again.
The hs troponin peaked at 14,143 ng/L. This is very high and is a typical level for an Occlusion MI [including STEMI (+) OMI].
Here is the post angiogram EKG:
All hyperacute T-waves are now normal
Here I put V4-V6 all on one image for the 5 ECGs
Now that hyperacute T-waves in V4-V6 become obvious. The ones on the far right are normal, after reperfusion.
There was no lesion that could be stented. She was treated medically for her presumed ACS MINOCA.
MINOCA
I do not have the bandwidth here to write a review of MINOCA.
But a few items:
The definition of MINOCA is predicated on the patient fulfilling all three main diagnostic criteria, namely:
1) the Universal Definition of Acute MI (which requires ischemia);
2) the presence of non-obstructive coronary artery on angiography (defined as no coronary artery stenosis ≥50%) in any potential infarct-related artery; and
3) the absence of another specific, clinically overt cause for the acute presentation.
MINOCA may be due to: coronary spasm, coronary microvascular dysfunction, plaque disruption, spontaneous coronary thrombosis/emboli, and coronary dissection; myocardial disorders, including myocarditis, takotsubo cardiomyopathy, and other cardiomyopathies.
We know that most type 1 acute MI due to plaque rupture and thrombosis occurs in lesions that are less than 50% (see Libby reference). This is in spite of the known proclivity of tighter stenoses to thrombose. The reason for this is population-based: there are many more moderate stenoses out in the population than there are tight stenoses, and so more MIs are generated from these moderate ones.
Even in patients whose moderate stenosis undergoes thrombosis, most angiograms show greater than 50% stenosis after the event. However, one can certainly imagine that many thromboses of non-obstructive lesions completely lyse and do not leave a stenosis on same day or next day angiogram. Coronary thrombosis with complete lysis is clearly possible, but its contribution to MINOCA is really not known because adequate investigation is rarely undertaken. The problem is difficult to study because angiographic visualization of arteries is not perfect, and not all angiograms employ intravascular ultrasound (IVUS) to assess for unseen plaque or for plaque whose rupture and ulceration cannot be seen on angiogram.
Furthermore, the clinical presentation of sudden chest pain, typical ECG findings of occlusion (hyperacute T-waves in this case), ECG findings in a coronary distribution, rise and fall of troponin with peak in the typical range for STEMI/OMI, and new wall motion abnormality in the area indicated by the ECG, must be considered to be due to coronary thrombosis. The degree of stenosis is not a great predictor of thrombosis, and culprits may not be visible. Even if there is a tight stenosis, it is not proof of culprit, as many individuals have tight fixed stenoses at baseline. There may be a chronic tight stenosis and a non-obstructed lesion that thrombosed.
Contemporary research studies of MINOCA have evaluated the prognosis of these patients, reporting a 12-month all-cause mortality of 4.7% (95% confidence interval, 2.6–6.9),3 with comparative studies consistently demonstrating a better prognosis than for those who experience AMI associated with obstructive coronary artery disease.
Lindahl et al. associated typical Myocardial Infarction therapies such as statins and ACE inhibitors with significantly decreased 1 year mortality in MINOCA patients, which suggests that they do indeed have a similar pathophysiology to MI patients with obstructive coronary disease.
From UpToDate:
Acute thrombosis at the site of non-obstructive eccentric plaque thrombosis — Many atherosclerotic plaques expand outward rather than encroaching on the arterial lumen. These ”positively-remodelled” plaques are often lipid rich and have a thin fibrous cap; they are vulnerable to rupture into the lumen [1,9,10]. Transient and partial thrombosis at the site of a non-obstructive plaque with subsequent spontaneous fibrinolysis and distal embolization may be one of the mechanisms responsible for the occurrence of MINOCA. Similarly, coronary erosion with loss of surface endothelium, possibly due to hyaluronan and neutrophil accumulation, can also cause MINOCA [1,11]. (See "Mechanisms of acute coronary syndromes related to atherosclerosis".)
The reason for these cases to be labeled as MINOCA is that angiography is of limited utility for the purpose of elucidating plaque-related thrombosis as a cause of thrombosis due to its low resolution as well as the fact that it does not interrogate the lumen of the vessel. Thus, intracoronary imaging modalities are crucial in this setting. Plaque rupture or erosion has been diagnosed by intravascular ultrasound in about 40 percent of women with MINOCA [12]. Optical coherence tomography, due to its high resolution, may provide additional information [10,13].
As MINOCA is associated with a risk of recurrent cardiovascular events over time, comparable with that of patients with acute coronary syndromes (ACS) and obstructive atherosclerosis [5,14,15], these patients require dual antiplatelet treatment for 12 months and statins. In particular, long-term lipid-lowering therapy with statins after MI is associated with a significant increase of the fibrous-cap thickness, paralleling the reduction of the lipid content of the plaque [16]. (See "Prevention of cardiovascular disease events in those with established disease (secondary prevention) or at very high risk".)
From Gue at al.
STEMI MINOCA versus NSTEMI MINOCA
STEMI occurs in the presence of transmural ischaemia due to transient or persistent complete occlusion of the infarct-related coronary artery. In patients presenting with non-ST-segment elevation MI (NSTEMI), the infarct is subendocardial. This pathophysiological difference also seems to be present within the MINOCA cohort. Registry data indicate that 6–11% of patients with acute MI have nonobstructive coronary arteries. Within the literature, MINOCA tends to present more commonly as NSTEMI than STEMI: the incidence of MINOCA reported in patients presenting with NSTEMI is about 8–10% and in STEMI cohorts it is 2.8–4.4%. This has resulted in an under-representation of STEMI MINOCA patients in the literature. Most studies examine undifferentiated ACS cohorts, with only a handful providing separate data. These studies indicate that the 1-year mortality of MINOCA presenting as STEMI is 4.5%, in contrast to the mortality of unselected MINOCA ACS patients who have a mortality of 4.7%. The underlying aetiology of MINOCA is similar among those presenting with STEMI and in all-comer MINOCA patients with ACS, with non-coronary aetiology responsible for presentation in 60–70% of individuals with STEMI and in 76% of unselected ACS patients.
References:
1. Lindahl B, Baron T, Erlinge D, et al. Medical Therapy for Secondary Prevention and Long-Term Outcome in Patients With Myocardial Infarction With Nonobstructive Coronary Artery Disease. Circulation [Internet] 2017;135(16):1481–9. Available from: http://dx.doi.org/10.1161/CIRCULATIONAHA.116.026336 https://www.ahajournals.org/doi/epdf/10.1161/CIRCULATIONAHA.116.026336
2. Pasupathy S, Tavella R, Beltrame JF. Myocardial Infarction With Nonobstructive Coronary Arteries (MINOCA): The Past, Present, and Future Management [Internet]. Circulation. 2017;135(16):1490–3. Available from: http://dx.doi.org/10.1161/CIRCULATIONAHA.117.027666 https://www.ahajournals.org/doi/epdf/10.1161/CIRCULATIONAHA.117.027666
3. Gue YX, Kanji R, Gati S, Gorog DA. MI with Non-obstructive Coronary Artery Presenting with STEMI: A Review of Incidence, Aetiology, Assessment and Treatment. Eur Cardiol [Internet] 2020;15:e20. Available from: http://dx.doi.org/10.15420/ecr.2019.13
4. Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med [Internet] 2013;368(21):2004–13. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23697515