This is another great case and discussion was contributed by Brooks Walsh, with some additions by Smith.
Here we discuss the differentiation of true positive from false positive ST elevation in the setting of LVH.
What do you think of this ECG in a patient with chest pain?
Case history
A middle-aged woman with a history of HTN, but no prior CAD, presented to the ED with chest pain. The pain had been mild and intermittent for 2 weeks, but had become more intense on the night of presentation. Her vitals signs were remarkable for marked hypertension. An ECG was obtained at triage:
Here we discuss the differentiation of true positive from false positive ST elevation in the setting of LVH.
What do you think of this ECG in a patient with chest pain?
 |
| There is ST elevation, but also high voltage (though the high voltage is NOT in the leads with worrisome STE, rather, it is in aVL). Is the ST elevation due to LVH? |
Case history
A middle-aged woman with a history of HTN, but no prior CAD, presented to the ED with chest pain. The pain had been mild and intermittent for 2 weeks, but had become more intense on the night of presentation. Her vitals signs were remarkable for marked hypertension. An ECG was obtained at triage:
ECG #1 at 0000 hours.
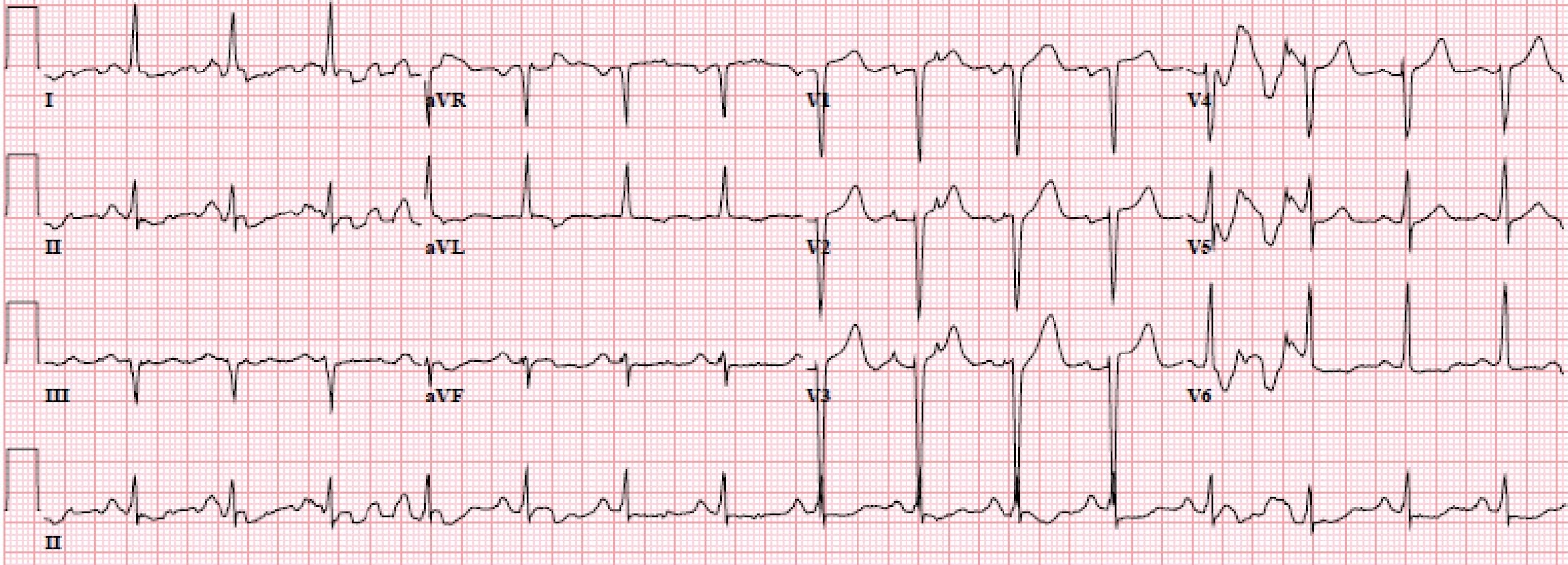 |
| If you ignore the baseline wander, there is non-significant STE in V1 and V2. The QRS in aVL varies in height, but one complex has an R-wave amplitude greater than 11 mm, which is very specific for LVH. The ST depression in V6, and T wave flattening/inversion in V6 and aVL, also support LVH |
The ECG was repeated to get a clearer tracing and because her pain increased to 10/10 at that moment.
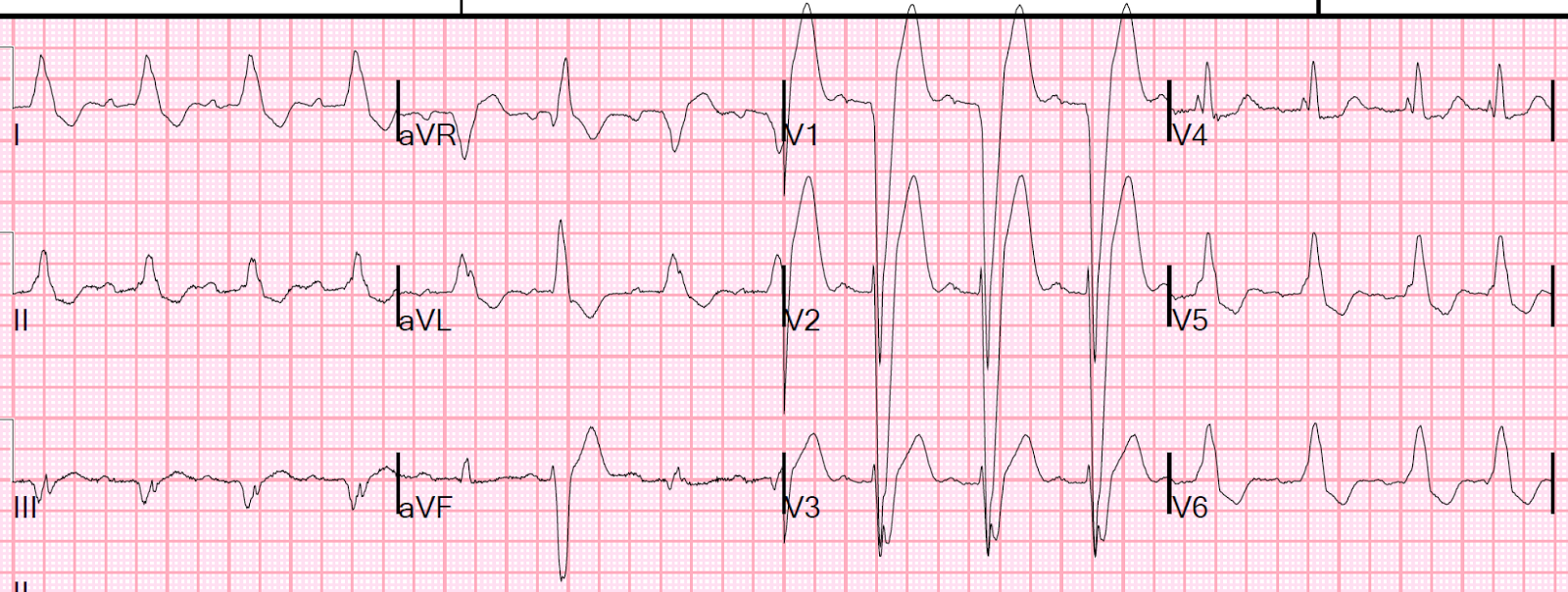
ECG #2 at 0004 hours (this is the one at the top of the post).
 |
| Only 4 minutes after ECG #1, there is greater STE in V1-V3, although it is concave-upwards. The T-waves in those leads have also significantly increased. There is ST depression in leads I, II, and aVL. The computer displayed an *** ACUTE AMI SUSPECTED*** alert. However, computers are often fooled by LVH. The criteria of Armstrong et al. would require the ST/S ratio to be 25% for diagnosis of STEMI in LVH. This is the same number used for the modified Sgarbossa criteria for LBBB. But here the ratio is only 14% - 18%, so those criteria would say that this is not LAD occlusion. |
The physician was concerned about STEMI, but also worried that she was overreacting, with the potential that LVH was producing a "STEMI-mimic."
In spite of those worries, she activated the cath lab.
A bedside echo was performed:
There is LVH, but also a septal wall motion abnormality.
No prior echocardiogram was available for comparison.
The ECG was repeated when the patient reported her pain had decreased.
ECG #3 at 0018 hours.
 |
| Similar to ECG #1, there is non-significant STE in V1- V2, and R in aVL greater than 11 mm. Also note the prolonged R-wave peak time in V5, V6, and the ST depression in lead I, and small amount of T-wave inversion in aVL. All these factors, again, support an ECG diagnosis of LVH |
The patient was nonetheless taken for emergency angiography, and a 99% mid-LAD lesion was found and stented.
ECG #4, 24 hours later
 |
| Anterior T-wave inversion, indicating reperfusion. Interestingly, few signs of LVH at this point. |
Can you diagnose an ACO (STEMI) when you also have LVH?
LVH can mimic an acute anterior coronary occlusion (ACO) on the ECG.1 Emergency physicians and cardiologists can have difficulty distinguishing these,4 and LVH is often cited as a cause of “false-positive” emergent angiography for ACO.5,6
A number of approaches have been suggested to differentiate the ECG of chronic LVH from that of a patient with a superimposed ACO, but none of them have proven satisfactory. For example, the ECG can be compared with older tracings. However, the ST segments in patients with LVH may show significant variation over time in the absence of ischemia.3 Some have also suggested that the typically asymmetric T wave inversion (TWI) of LVH might be distinguished from the typically symmetric TWI of cardiac ischemia. However, no evidence is cited to support this.3
Is the STE concave or convex?
LVH usually has concave-upwards ST segments, but conVEX-upwards can also be seen, e.g. in these cases from Dr Smith’s book: The ECG in Acute MI:
 |
| Case 22-1. Chest pain in known LVH, negative troponins and stress test, no wall motion abnormality. |
 |
| Case 22-4. Received fibrinolytics twice, but negative troponins both times. Echo showed LVH only, no wall motion abnormality. Note also that the T-waves are not tall! |
Conversely, an ACO of the LAD will usually produce STE in the anterior leads with convex-upwards ST segments. Smith, however, showed that many ACOs of the LAD (without LVH) had conCAVE-upwards morphology on the initial ECG.8
(This was likely a function of time: patients with a shorter
time between symptom onset and ECG were less likely to show convexity.)
An example of acute LAD occlusion with upwardly concave ST segments in every lead of V2-V6:
 |
| Note again that the T-waves are not tall |
Is there a “Smith-modified-Sgarbossa” rule for ST elevation in LVH?
Similar
to the anterior STE seen with LBBB, the degree of STE in leads V1-V3 of
patients with LVH is generally proportional to the depth of the
proceeding S wave. In the case of LBBB, it has been shown that STE that exceeds 25% of the proceeding S-wave depth is disproportionately high, and identifies ACO with excellent specificity.9 Could this also work in LVH?
One retrospective analysis by Armstrong et al. suggested that, with LVH, STE in V1-V3 that exceeds 25% of the preceding QRS complex could be an accurate means for ruling out ACO, and fairly sensitive for identifying
ACO.
Smith comment: The Armstrong paper did not have appropriate methods to study this. The appropriate methods would be to take consecutive ECGs with high voltage and ST elevation in the leads with ST elevation, separate them into those with and without LAD occlusion, and see what are the differences in ST/S ratio. I have inserted at the bottom of this post some examples from Armstrong's paper. You will see that they are not cases that you would have difficulty with. I have tried to study this topic twice and failed because there are very few cases of high voltage in V1-V4 and LAD occlusion. In fact, even this case does not fit, as the voltage in the affected leads does not meet LVH criteria!
Most importantly, since STE in LVH rarely exceeds 4 mm in height, the 25% criterion is likely far too insensitive. For example, in a patient with an S-wave 30 mm in depth, the STE would have to exceed almost 7 mm.
Smith comment: The Armstrong paper did not have appropriate methods to study this. The appropriate methods would be to take consecutive ECGs with high voltage and ST elevation in the leads with ST elevation, separate them into those with and without LAD occlusion, and see what are the differences in ST/S ratio. I have inserted at the bottom of this post some examples from Armstrong's paper. You will see that they are not cases that you would have difficulty with. I have tried to study this topic twice and failed because there are very few cases of high voltage in V1-V4 and LAD occlusion. In fact, even this case does not fit, as the voltage in the affected leads does not meet LVH criteria!
Most importantly, since STE in LVH rarely exceeds 4 mm in height, the 25% criterion is likely far too insensitive. For example, in a patient with an S-wave 30 mm in depth, the STE would have to exceed almost 7 mm.
 |
| In our case, Lead V3 in ECG #2 shows STE/QRS = 3-4/22-24 for a maximum ratio of 17%, far short of the 25% criterion. Yet the LAD was occluded, or nearly so, at this time. |
Use the T wave, not the ST segment?
It is unclear if ischemia modifies T wave height or morphology in LVH.3 In our case, the dynamic rise and fall of the anterior T waves is striking, suggesting hyperacute T waves.10
 |
| See all three compared. The middle one is during LAD occlusion. |
However,
it has been difficult to define hyperacute T-waves in patients with
normal baseline ECGs, let alone those with LVH. Unfortunately, while
some authors have suggested that certain criteria (T-wave amplitude/QRS amplitude greater than 75%) could differentiate ACO from LVH,11 their cited study excluded patients with LVH.10
Is LVH like left ventricular aneurysm?
The repolarization behavior of LVA is likely distinct from that of LVH. However, an analogous rule for identifying superimposed ACO of the LAD in the presence of persistent STE after prior MI (aka “left ventricular aneurysm,” or LVA) was just validated.12 An acute MI is likely if the T-wave amplitude to QRS amplitude ratio exceeds 0.36 in any of leads V1-V4 when a patient has ECG signs of LVA.
It’s unclear if we can use the same analysis in patients with LVH. However, it is clear that, in lead V3 in Figure 2, the Tamp/QRSamp exceeds 0.36 by a significant margin. (It also does so in lead V2, at 7/14 = 50%).

Conclusion
It
is complete conjecture at this point, but perhaps we should be
measuring T-wave heights more often in suspected ACO. Close scrutiny of
T-waves have anecdotally been useful when no diagnostic ST elevation is seen, and even when a ventricular pacer has obscured the ST segment.
Illustration of why Armstrong's paper does not help us with the question at hand:
These two ECGs are from Armstrong's paper (5) and are supposed to illustrate how the ST/S ratio of greater than 25% works to differentiate LVH from STEMI
Illustration of why Armstrong's paper does not help us with the question at hand:
These two ECGs are from Armstrong's paper (5) and are supposed to illustrate how the ST/S ratio of greater than 25% works to differentiate LVH from STEMI
 |
| You can see that, in neither of these cases of anterior STEMI would you be asking whether the ST elevation is due to LVH or not.A rule that is useful would help you differentiate the ST elevation due to LVH from that which is due to STEMI superimposed on LVH. |
References
1. Huang HD, Birnbaum Y. ST elevation: differentiation between ST elevation myocardial infarction and nonischemic ST elevation. J Electrocardiol. 2011;44(5):494.e1-e494.e12. doi:10.1016/j.jelectrocard.
2. Brady
WJ, Chan TC, Pollack M. Electrocardiographic manifestations: patterns
that confound the EKG diagnosis of acute myocardial infarction—left
bundle branch block, ventricular paced rhythm, and left ventricular
hypertrophy1. J Emerg Med. 2000;18(1):71-78. doi:10.1016/S0736-4679(99)
3. Birnbaum Y, Alam M. LVH and the diagnosis of STEMI - how should we apply the current guidelines? J Electrocardiol. 2014;47(5):655-660. doi:10.1016/j.jelectrocard.
4. Brady
WJ. Electrocardiographic left ventricular hypertrophy in chest pain
patients: Differentiation from acute coronary ischemic events. Am J Emerg Med. 1998;16(7):692-696. doi:10.1016/S0735-6757(98)
5. Armstrong
EJ, Kulkarni AR, Bhave PD, et al. Electrocardiographic Criteria for
ST-Elevation Myocardial Infarction in Patients With Left Ventricular
Hypertrophy. Am J Cardiol. 2012;110(7):977-983. doi:10.1016/j.amjcard.2012.05.
6. Shamim
S, McCrary J, Wayne L, Gratton M, Bogart DB. Electrocardiograhic
findings resulting in inappropriate cardiac catheterization laboratory
activation for ST-segment elevation myocardial infarction. Cardiovasc Diagn Ther. 2014;4(3):215-223. doi:10.3978/j.issn.2223-3652.
7. Schocken DD. Electrocardiographic left ventricular strain pattern: Everything old is new again. J Electrocardiol. 2014;47(5):595-598. doi:10.1016/j.jelectrocard.
8. Smith SW. Upwardly concave ST segment morphology is common in acute left anterior descending coronary occlusion. J Emerg Med. 2006;31(1):69-77. doi:10.1016/j.jemermed.2005.
9. Meyers
HP, Limkakeng Jr. AT, Jaffa EJ, et al. Validation of the modified
Sgarbossa criteria for acute coronary occlusion in the setting of left
bundle branch block: A retrospective case-control study. Am Heart J. 2015;170(6):1255-1264. doi:10.1016/j.ahj.2015.09.005.
10. Collins MS, Carter JE, Dougherty JM, Majercik SM, Hodsden JE, Logue EE. Hyperacute T-wave criteria using computer ECG analysis. Ann Emerg Med. 1990;19(2):114-120. doi:10.1016/S0196-0644(05)
11. Sovari AA, Assadi R, Lakshminarayanan B, Kocheril AG. Hyperacute T wave, the early sign of myocardial infarction. Am J Emerg Med. 2007;25(7):859.e1-e859.e7. doi:10.1016/j.ajem.2007.02.
12. Klein
LR, Shroff GR, Beeman W, Smith SW. Electrocardiographic criteria to
differentiate acute anterior ST-elevation myocardial infarction from
left ventricular aneurysm. Am J Emerg Med. 2015;33(6):786-790. doi:10.1016/j.ajem.2015.03.
13. Neuman
Y, Cercek B, Aragon J, et al. Comparison of frequency of left
ventricular wall motion abnormalities in patients with a first acute
myocardial infarction with versus without left ventricular hypertrophy. Am J Cardiol. 2004;94(6):763-766. doi:10.1016/j.amjcard.2004.05.