Written by Pendell Meyers with edits by Smith
A man in his late 40s with no known medical problems was at work when he suddenly experienced midsternal chest pain radiating down both arms. Approximately 1 hour after onset of symptoms he was triaged at the ED, with ongoing chest pain, normal vitals, and this triage ECG:
 |
| What do you think? |
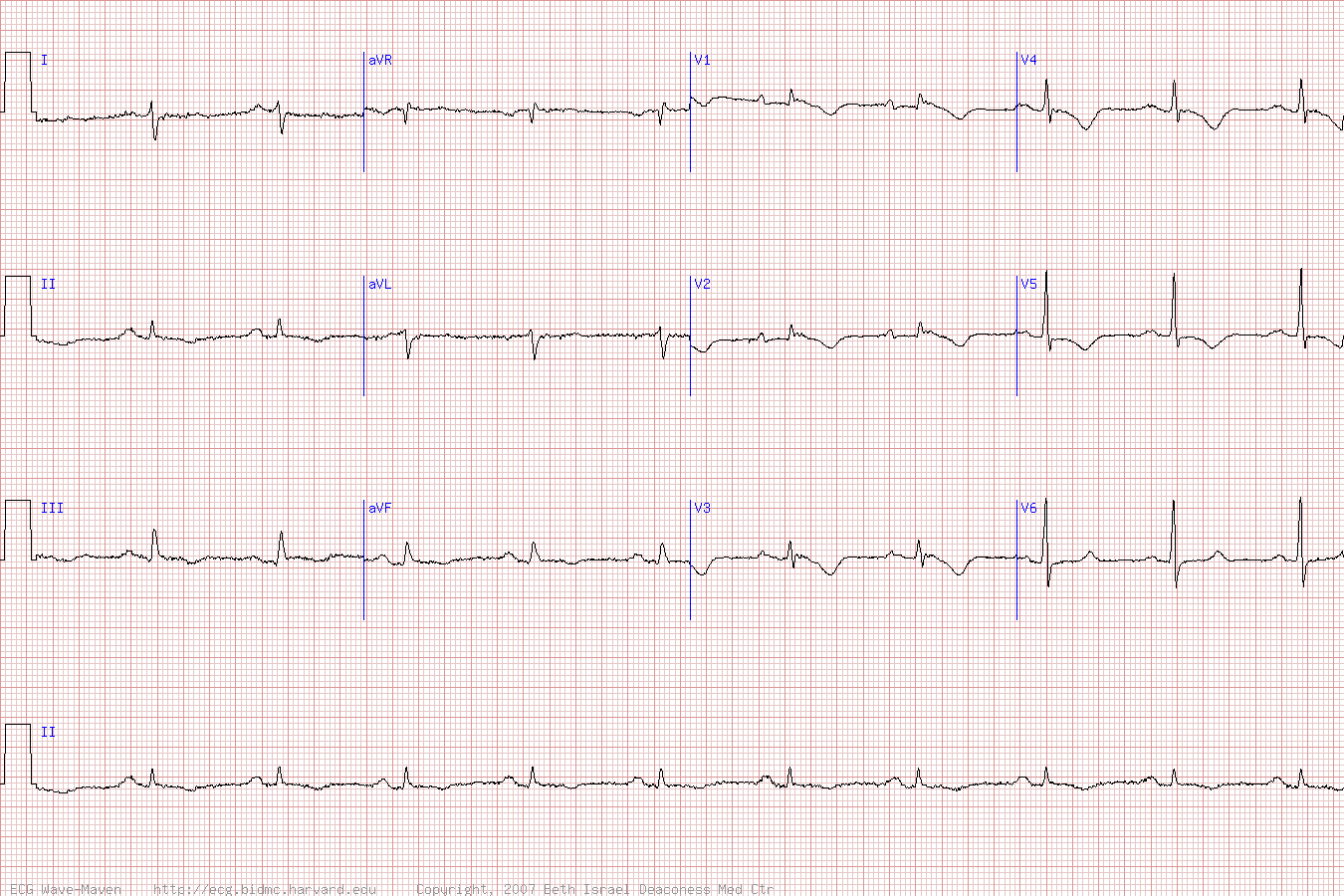
Twice, months apart, I sent this ECG to Dr. Smith without any context or other information (I do this many times per day, with many normal or false positive cases mixed in). The first time he responded "acute ischemia but not active occlusion". The second time he responded "LVH and subendocardial ischemia." It is the same response each time: active subendocardial ischemia.
The computer interpretation states "Left ventricular hypertrophy with repolarization abnormality." So it states there is an abnormality, but attributes it to LVH.
There is sinus rhythm with normal QRS (except for borderline high voltage and LAD, some would call it LAFB - LVH can mimic LAFB). There is a tiny but definite hint of STD in V4-V6, maximal in V5 and V6. There is a tiny amount of STD in I and II and aVL. There is a tiny amount of reciprocal STE in aVR. There is a tiny amount of STE in V1. With such a normal QRS complex, none of these ST segment changes are explained by the QRS complex, and all of them must be considered new and significant until proven otherwise.
In this pattern, with STD maximal in V5-6 with reciprocal STE in aVR, the ECG is diagnostic for supply/demand mismatch ischemia, also known as subendocardial ischemia (as opposed to OMI, or focal subepicardial ischemia). The ECG pattern of supply/demand ischemia is largely identical regardless of the cause, whether or not it is due to decreased oxygen supply (e.g. left main ACS but with collaterals or without total occlusion, triple vessel disease with non-occlusive ACS, decreased blood pressure, hypoxemia, etc.), increased demand (infinite number of non-ACS causes), or both.
Thus, the interpretation of the supply/demand mismatch pattern on ECG depends completely on the clinical context.
This patient's clinical context is clearly most likely ACS. Therefore the interpretation is acute, active, non-occlusive ischemia until proven otherwise. This could be compatible with left main ACS (but not total occlusion without collateral circulation) or basically any other acute non-occlusive ACS event causing a significant area of supply/demand mismatch.
So the patient has definite ACS symptoms, with an ECG showing active ongoing ischemia. He should be assumed to have an acute culprit lesion which could fully occlude at any moment, or could cause significant ischemia and complications at any moment even without fully occluding.
Below you can see close-ups of leads V5, V6, and aVR, with and without red lines showing the baseline, highlighting the small but definite and very important STD and STE.
Case Continued:
The ECG was read as sinus rhythm with no signs of STEMI (that's of course true). Due to overcrowding, and thinking that he had normal vital signs and a "normal" ECG, he had initial labs drawn and was sent to the waiting room until a regular room became available. No medicines were given at that point.
Approximately 30 minutes later, he called out for help in the waiting room. He vomited and collapsed, and was found to be in cardiac arrest. CPR was started and he was brought to the resuscitation area where his rhythm was described as "fine VFib." Multiple defibrillation attempts were made, as well as continuous high quality CPR, but there was no change in rhythm, never any brief ROSC. Thrombolytics were considered but not given due to concern that the etiology was not clear, and they stated they were considering aortic dissection as a possible cause. The arrest was called after about 40 minutes.
The troponin drawn at triage resulted at 121 ng/L (upper reference limit 20 ng/L for men). It is unclear whether the team had this information during resuscitation.
An autopsy was later performed, showing no PE or dissection, but an 80% stenosis of the proximal left circumflex artery. The report does not comment on whether there were signs of fresh thrombus at this location, or whether they could tell if there were definitively acute plaque rupture. There was also 50% luminal stenosis of the mid RCA.
The autopsy report states:
"Myocardial infarcts less than four hours old show no histologic changes so it is difficult to prove an acute myocardial infarct in less than that time period. Correlating the patient's substernal chest pain and coronary artery disease with cardiac arrest and inability to return to sinus rhythm, a myocardial infarct is the presumed cause of death. Pertinent negatives include that there was no evidence of aortic rupture or dissection. There was no pulmonary embolus. There were minimal pathologic changes throughout the body."
We did not need an autopsy to conclude that ACS was the most likely cause of death, but this autopsy obviously supports that theory. It seems overwhelmingly likely to me that the LCX lesion was the acute culprit. It could have caused VF arrest without fully occluding, or it could have fully occluded in the waiting room and then caused VF arrest (without any ECG performed after triage). One third of patients with confirmed STEMI on the ECG do not have 100% occlusion (TIMI 0 flow) at the time of their emergent cath, so it is very likely that a full LCX occlusion could be only 80% at the time of autopsy. But again, one need not have occlusion for ischemia to result in VF arrest.
Learning Points:
1. Any ischemia can result in ventricular fibrillation, even non-occlusive ischemia.
2. Moreover, non-occlusive thrombi can become occlusive: thrombi are dynamic; they propagate and lyse continuously.
3. Patients with symptoms that have even just a moderate likelihood of being due to ACS or due to any ongoing ischemia should not be sent to the waiting room. They need to be on a monitor in case of dysrhythmia, and take priority over most other patients in the ED.
4. Even if you are not certain that ST depression represents ischemia, it is prudent to at least have the patient in a monitored bed while you are undertaking more investigation. These patients should be a "stat placement" to monitored bed.
Commentary
There is confusion created by the educational campaign of "ST elevation in aVR." Often, the first thought that providers have when they see STE in aVR is that the patient has "left main occlusion," even in a clinical context that is not consistent with ACS. And yet when this patient with clear clinical ACS presents with1 mm of reciprocal STE in aVR, it was not seen or understood. We have a significant number of unnecessary cath lab activations due to STE in aVR because providers do not understand that it is one of the most common ECG findings in patients who have any significant illness causing supply/demand mismatch. This is why I title my aVR lecture: "Lead aVR: Once "forgotten," now remembered, always misunderstood." It's gotten to the point that cardiology perceives that we are crying wolf. This is one of the most common findings other than obvious STEMI that cardiologists get consulted for, but the majority of the time it is simply supply/demand mismatch due to a variety of etiologies, including AF with RVR, hypovolemia, GI bleed, respiratory failure, sepsis with hypotension, aortic stenosis, and others.
STD maximal in V5-6 and lead II, with reciprocal STE in aVR, indicates global supply/demand mismatch subendocardial ischemia, which can be due to ACS or non ACS causes.
It is important to realize that ischemic ST depression due to subendocardial ischemia does not localize to the leads which show ST depression. There is usually an ST depression vector towards the apex of the heart (leads II and V5, V6), with reciprocal STE in aVR, and if it is very profound, it is often due to left main or LAD ischemia, or due to non-occlusive ACS of any vessel along with disease of all 3 vessels. However, non-occlusive ischemia of any artery can result in such ischemic ST depression even in the absence of 3 vessel disease.
STD maximal in V1-V4, in contrast to V5-V6, is reciprocal to subepicardial ischemia due to OMI (reciprocal to what would manifest as ST elevation of overlying leads if they were there). The exceptions are when there is an abnormal QRS to account for secondary STD, such as RBBB or to a non-ischemic etiology of STD such as hypokalemia.
Stay tuned for our upcoming publication in JAHA on STD maximal in V1-V4 vs. V5-V6.
Aspirin should be given at triage for a clinical history and ECG like this, unless there is some strong suspicion of dissection (not present in this case). Dissection is far less common than ACS.
After a VF arrest, there should be no doubt that this was ACS and that coronory thrombosis was the etiology. When the patient could not be resuscitated, this patient would have been a prime candidate for ECMO. If he had arrested in cities such as Minneapolis, with refractory VF ECMO programs (before the program was suspended due to Covid), he would likely have been an ideal candidate for ECMO, in a population that seems to be receiving a nearly 50% chance of neurologically intact survival. If ECMO is not an option, and going to the cath lab during arrest is not an option, I would try thrombolytics before ending the resuscitation on a young healthy witnessed ACS arrest.
Transesophageal echo (TEE) can help decide between asystole and fine VF.
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}