A 50-something patient who had just had a median sternotomy called 911 for chest pain.
The medics put him on the monitor and saw this:
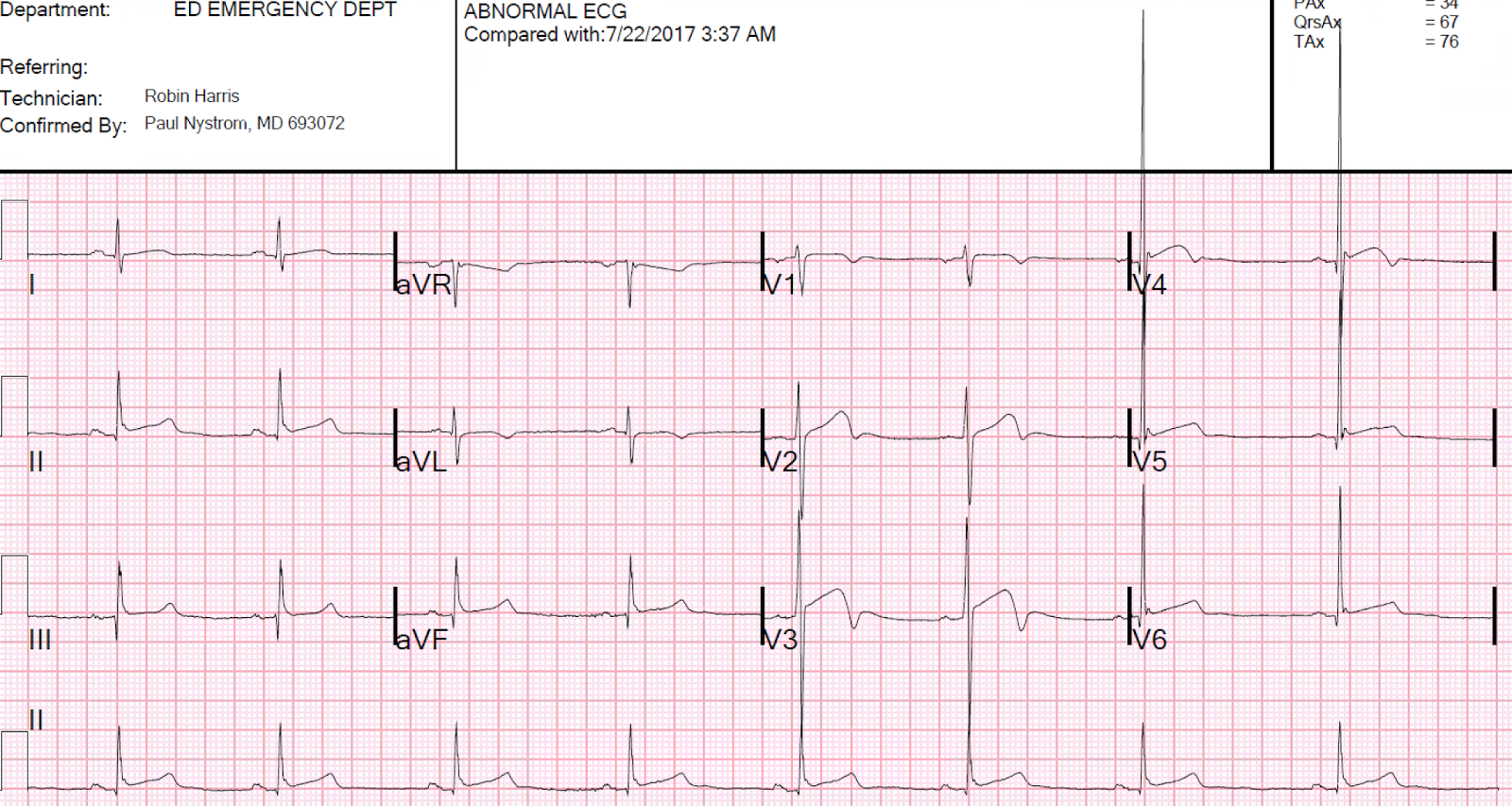
This was worrisome to them. They always record a 12-lead, and did so this time:
On arrival, I viewed this ECG and immediately de-activated the cath lab. Even without viewing an old ECG, one can know that this ST elevation is entirely secondary to LVH. The morphology is classic and you just have to learn to recognize it. There is a deep S-wave in each of V1-V3, so the ST elevation is abnormal repolarization that is a result of abnormal depolarization: it is "secondary" ST elevation, not "primary."
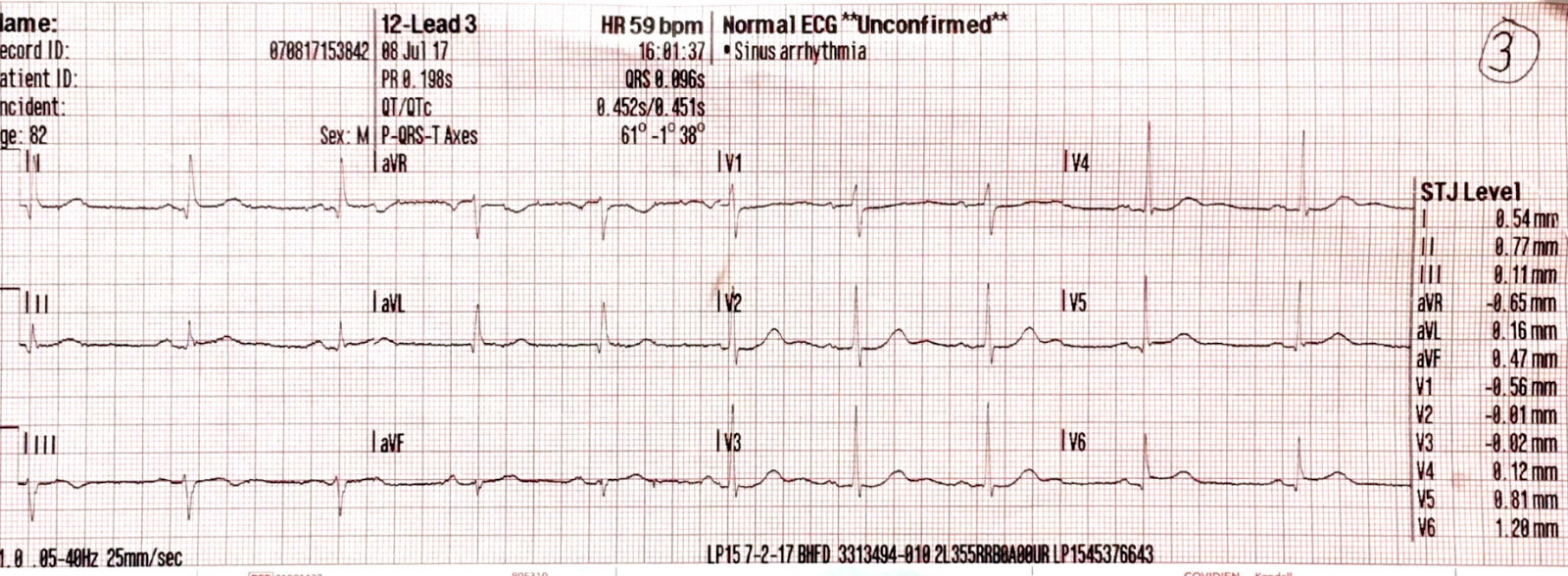
We recorded another ECG in the ED:
How about that rhythm strip?
1. The S-wave is cut off on the rhythm strip, so the ST elevation cannot be interpreted in the context of the QRS, as it must be!
2. Rhythm strips use different filtering techniques than 12-lead ECGs. Suffice it to say that you cannot trust ST changes on monitor strips. It is fine if they lead you to obtain a 12-lead ECG, but they should never be trusted by themselves, as they are very often misleading.
Learning Points:
1. Learn to recognize the ST elevation that is the result of LVH. See this detailed post on the topic:
The medics put him on the monitor and saw this:
 |
| Note the apparent massive ST Elevation in what is lead MCL1 (similar to V1) What do you think? |
This was worrisome to them. They always record a 12-lead, and did so this time:
 |
| There is a large amount of ST elevation in V1-V3. The medics considered this diagnostic of STEMI and activated the cath lab. What do you think? |
On arrival, I viewed this ECG and immediately de-activated the cath lab. Even without viewing an old ECG, one can know that this ST elevation is entirely secondary to LVH. The morphology is classic and you just have to learn to recognize it. There is a deep S-wave in each of V1-V3, so the ST elevation is abnormal repolarization that is a result of abnormal depolarization: it is "secondary" ST elevation, not "primary."
We recorded another ECG in the ED:
 |
| This was identical to the patient's previous ECG. |
How about that rhythm strip?
1. The S-wave is cut off on the rhythm strip, so the ST elevation cannot be interpreted in the context of the QRS, as it must be!
2. Rhythm strips use different filtering techniques than 12-lead ECGs. Suffice it to say that you cannot trust ST changes on monitor strips. It is fine if they lead you to obtain a 12-lead ECG, but they should never be trusted by themselves, as they are very often misleading.
Learning Points:
1. Learn to recognize the ST elevation that is the result of LVH. See this detailed post on the topic:
LVH with anterior ST Elevation. When is it anterior STEMI?
2. ST segments on cardiac monitors cannot be trusted, and must be verified by a 12-lead ECG.