An otherwise healthy woman in her 20's presented with tachycardia. She had experienced palpitations and called 911. Prehospital rhythm strips were at a rate of at least 200 (unavailable) and the medics gave adenosine at both 6 mg and 12 mg with no effect. She was very stable with no CP, SOB, hypotension or evidence of shock.
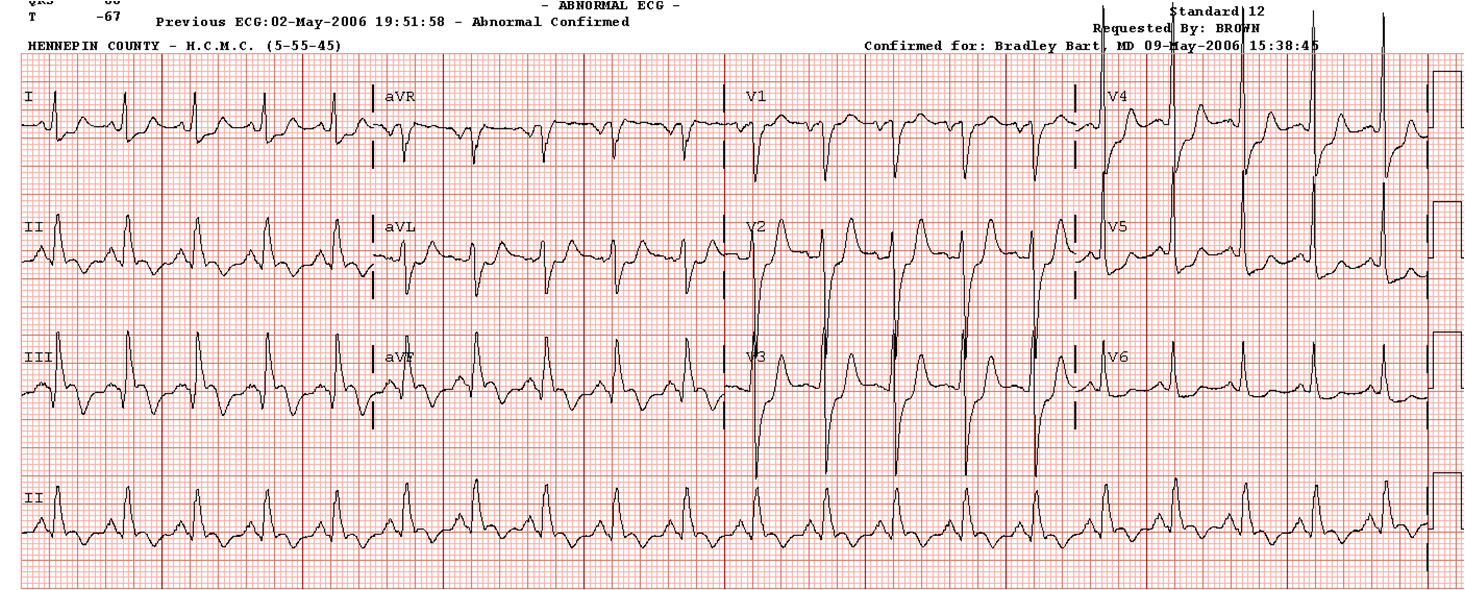
Here is the initial ED ECG:
 |
What is the diagnosis (this is pathognomonic)? See below.
(Notice that the computer incorrectly read ***Acute MI***)
|
1. The rhythm is irregularly irregular, therefore it is atrial fibrillation
2. The complexes are wide (so one might think of atrial fibrillation with aberrancy, in which case you should see RBBB or LBBB pattern, which is not there)
3. It is very fast (200 bpm)
4. The shortest R-R interval (between complexes 12 and 13) is about 240 ms (very short)
5. The complexes look bizarre and are not uniform, as they would be with simple aberrancy. Thus, these represent differentially pre-excited ventricular myocardium.
This is atrial fibrillation in the setting of WPW, and is a dangerous rhythm which can degenerate into ventricular fibrillation. It is more likely to degenerate if the physicians gives AV nodal blocking drugs, especially calcium channel blockers.
Here is another example of this.
How should this be managed? It can be managed with medications
that convert atrial fibrillation to sinus, such as procainamide or
ibutilide (and others), but when you have a wide complex very fast
tachycardia,
it is best to use electrical cardioversion. It is the safest, and keeps
you from having to make a definite diagnosis.
As long as you can manage procedural sedation, which is very easy in the case of cardioversion because you only need seconds of sedation and amnesia, then cardioversion is the safest method.
The pattern was not recognized as pre-excitation, but as atrial fib with aberrancy. These were very smart and very experienced physicians, but anyone can make a snap judgment followed by premature closure,
and this is another reason why electricity is the safest - you don't need to have the correct diagnosis). Subsequently, the physicians gave the patient first diltiazem and then esmolol, with no ill effect but also little beneficial effect. It could have resulted in degeneration to ventricular fibrillation. AV nodal blockade is particularly dangerous when there is a shortest R-R interval of less than 250 ms (as here), and especially if less than 220 ms. Adenosine is also contraindicated.
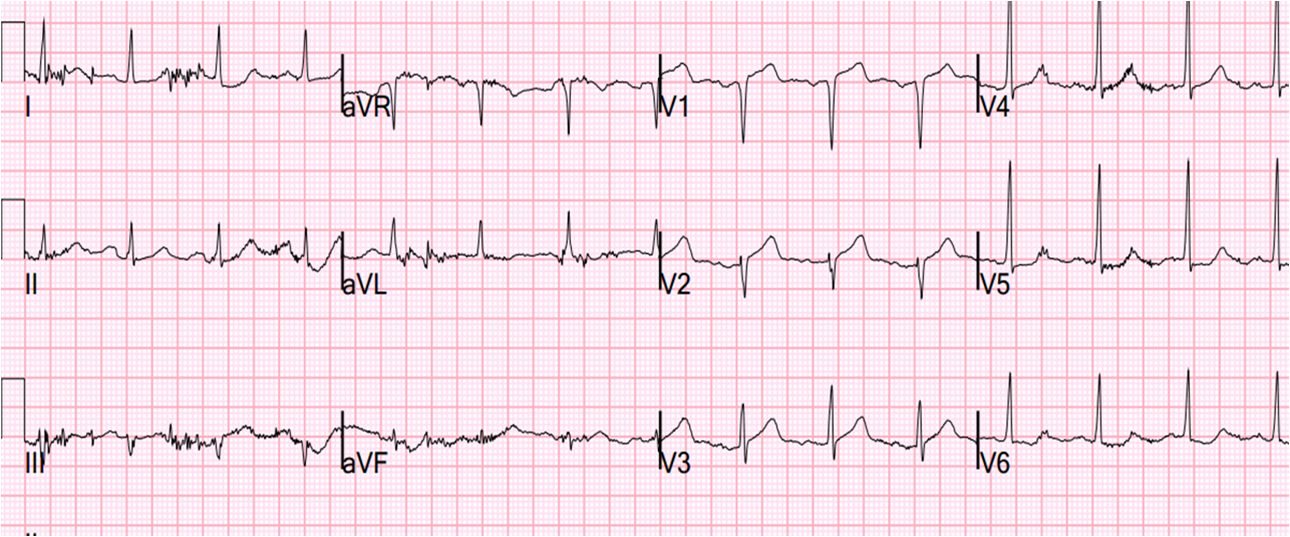
After dialing up the esmolol, the patient spontaneously converted to NSR and had the following ECG:
After conversion:
 |
| Sinus rhythm with short PR interval and large delta waves seen best in precordial leads, and confirming Wolff Parkinson White (WPW) syndrome of pre-excitation down an accessory pathway. There can be positive, negative, or isoelectric delta waves depending on their own axis. WPW may greatly change both depolarization (in this case with large upright R-waves in right precordial leads because the accessory pathway is left lateral, depolarizing the ventricle from left to right) and repolarization (see these cases for acute MI mimics due to WPW) |
Followup, and what is "Concealed Conduction"?
Her charts revealed 3 previous visits for palpitations, and in all cases the ECG was interpreted as normal. Here is one of them:
 |
There are very subtle delta waves which I do not believe I would have noticed propectively, but in retrospect look to be real. The computer read a QRS duration of 106 ms which is borderline long and due to these subtle delta waves. The PR interval is normal because the delta waves are so minimal.
|
|
This is
concealed conduction, or nearly concealed. That is to say, the presence of an accessory pathway is not evident (at least to most observers) on the baseline surface ECG. (There are times when it is truly concealed, and there are no delta waves even in retrospect). It is important to know about concealed conduction so that if you suspect
WPW as a cause of tachycardia that is now resolved, you will not rule out the diagnosis by a normal baseline ECG.
There are two mechanisms of concealed conduction:
1. Conduction through the accessory pathway is retrograde only (mechanism unclear)
2. The impulse reaches the AV node and gets through to the ventricles
before it gets to, and through, the accessory pathway.
The second mechanism applies in this case: Look at the first two ECGs above (those with abnormal conduction). Notice that the R-wave in V1 is very large, as it would be in RBBB. This is because the impulse is going down a left lateral bypass tract and then proceeding through the myocardium from left to right, resulting in a large R-wave in V1. Thus, the bypass tract (accessory pathway) is to the left lateral of the left atrium, which is far from the sinus node (right part of right atrium). When the AV node is conducting fast (such as with anxiety, low vagal tone, high catecholamines, etc.), then the impulse gets to and through the AV node and through the Purkinje system before it makes it down the accessory pathway and therefore there is no (or minimal) delta wave. On the other hand, if the AV node conduction is slower, then the delta waves will be evident.
However, even in WPW with concealed conduction, the accessory pathway is always available to cause trouble!
In both types of concealed conduction, it can result in orthodromic reciprocating supraventricular tachycardia (re-entrant down through the AV node and up through the bypass tract), and this cannot be differentiated from intranodal reentrant (standard) SVT on the surface ECG.
In the second type of concealed conduction, it can result in
three abnormal rhythms:
First, there can be orthodromic re-entrant reciprocating tachycardia.
Second, there can also be
antidromic re-entrant reciprocating tachycardia (which is a regular wide complex tachycardia). And
third, if atrial fibrillation develops, then it will manifest as this dangerous wide complex tachycardia.
Why did she not have concealed conduction on the post conversion ECG? --Because she had received AV nodal blockade with diltiazem and the pathway down the AV node was slow.


























{kind=link}