Brooks Walsh @BrooksWalsh helped with this post
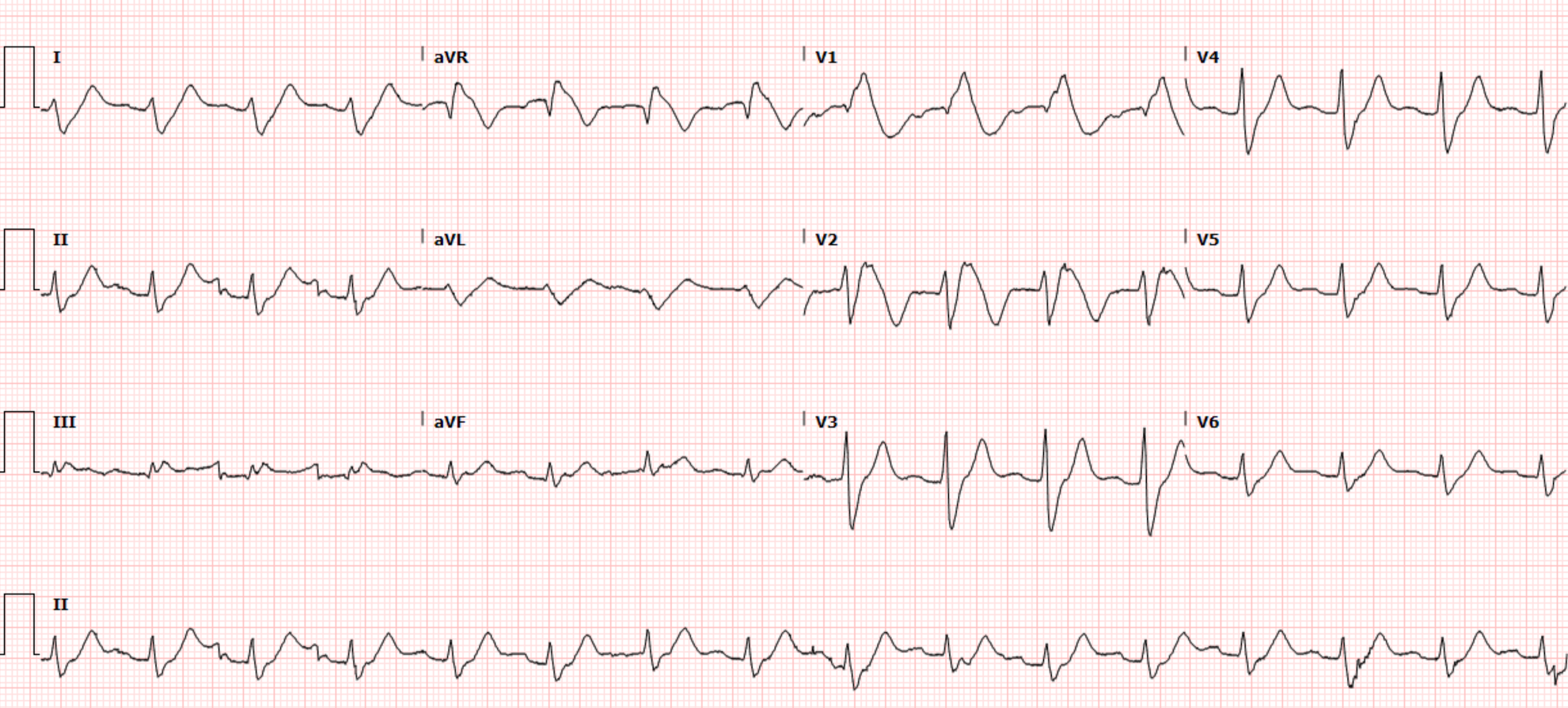
One of my partners showed me this series of ECGs, without any info:

I said: "It's a normal variant. Young black male, right?"
He said, "Yes, but look at this one recorded 2 hours later. It is different":

I said: "Yes, small changes can happen even with normal variants." And there might be a slight difference in lead placement. On the 2nd ECG, V4 is farther to the right -- notice there is more S-wave than on the first and the R/S ratio is smaller.
There was also a 3rd ECG at 3.5 hours:

I said "So tell me about the patient. Chest pain? Young black male?"
"Yes, 33 yo black male with acute chest pain."
Then I said, "I would just rule him out with troponins."
He said: "He had 2 hs troponin I less than 4 ng/L."
I said, "Then that is all you need."
He said, "Well, I called cardiology and got a formal echocardiogram. It was totally normal. How did you know?"
I said, "First, there is very high voltage, probably because he has thin chest wall and, second, I recognize this morphology. Some things simply require pattern recognition."
Comments
T-wave inversion, with or without ST Elevation, in young black males, especially athletes, is very complicated, but not so much because it mimics ACS, but because it is difficult to ascertain if the ECG represents Hypertrophic Cardiomyopathy.
If the patient presents with chest pain, and MI/ACS is the main concern, then you can recognize these patterns as NOT due to ACS. The patient then does NOT need further ED workup.
If the patient presents with syncope, then HOCM, or even Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) is on the differential and it becomes much more difficult to differentiate the benign forms from HOCM, and an echocardiogram may be necessary, and may even be necessary before sending the patient home. Perhaps a POCUS cardiac ultrasound is sufficient, if the operator/interpreter knows what he/she is doing.
Here is a post/lecture on Benign T-wave Inversion, with several example ECGs: http://hqmeded-ecg.blogspot.com/2012/03/benign-t-wave-inversion.html
Here is an article that Brooks and I wrote, along with Peter MacFarlane:
Distinctive ECG patterns in healthy black adults.
Full text manuscript: https://eprints.gla.ac.uk/189824/7/189824.pdf
The below quote is from this article:
Electrocardiographic anterior T-wave inversion in athletes of different ethnicities: differential diagnosis between athlete's heart and cardiomyopathy
Here is full text (images at this article are worth viewing)
"Older studies found that anterior TWI was found in 3%–10% of black Africans or African Americans [30,40,65,67], but not in white subjects [65]. More contemporary literature finds that anterior TWI can be found in 4.2% of healthy, non-athletic black males [82]. By contrast, anterior TWI is seen in only 1.9% of white male athletes [82], and rarely extends
past V2 [83]. The prevalence in non-athletic middle aged white men is even lower (0.5%) [72]. Similarly, this pattern is found in 14% of black European female athletes, but only 2% of white female athletes [71].This pattern was found in over 12% of black European male athletes [82], and is recognized as a benign feature in the black athlete [84]. This may be true in athletes of any race: one study has demonstrated that the combination of J point elevation N0.1mV and TWI limited to V1-4 ruled out arrhythmogenic right ventricular cardiomyopathy and hypertrophic cardiomyopathy, regardless of ethnicity [85]. In the absence of J point elevation, HCM or ARVC must still be considered. The literature hasn't
specifically addressed whether this pattern of anterior TWI with J-point elevation can be presumed benign in non-athletes, black or white."


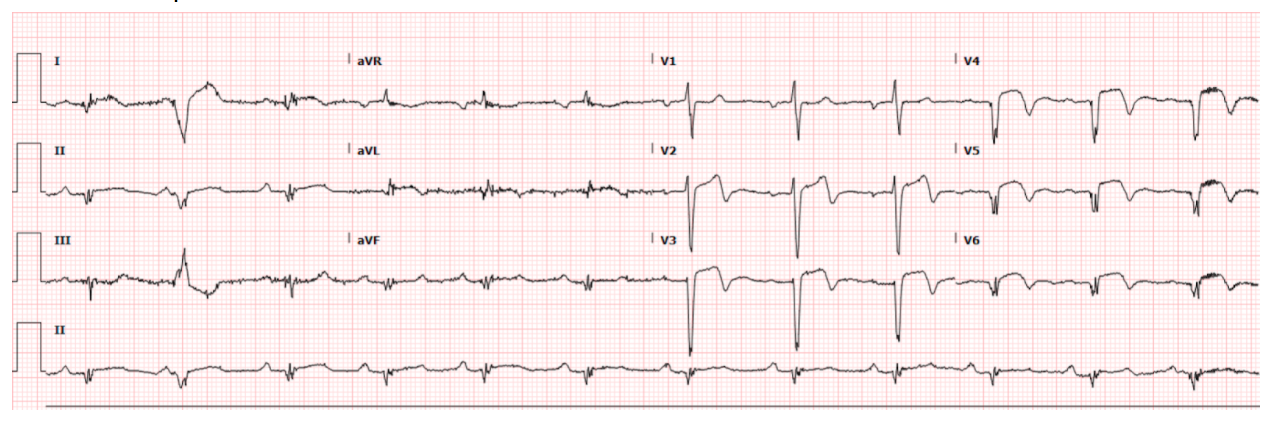
Below are 2 representative tracings from HOCM patients. Note that there is no J-point elevation. However, I believe these are very different from our patient because there is poor R-wave progression. Our patient above has very well formed R-waves (and S-waves) in V3 and V4.

Bottom line:
If chest pain, recognize this pattern as NOT OMI and NOT Wellens'. Do get serial troponins to rule out Non-OMI.
If syncope, beware HOCM, and much less commonly, ARVC.